
Газета «Новости медицины и фармации» Гастроэнтерология (304) 2009 (тематический номер)
Вернуться к номеру
Болезнь Вестфаля — Коновалова — Вильсона
Авторы: О.А. Голубова, Донецкий национальный медицинский университет
Версия для печати
Болезнь Вестфаля — Коновалова — Вильсона (син. гепатоцеребральная, гепатолентикулярная дистрофия или дегенерация) — редкое генетическое заболевание с аутосомно-рецессивным типом наследования, обусловленное нарушением обмена меди в организме. На начальных стадиях болезни медь накапливается в печени, что приводит к развитию гепатита, цирроза или фульминантной печеночной недостаточности. На последующих этапах медь накапливается в других органах и системах, преимущественно в центральной нервной системе и почках, также вызывая их повреждение.
Метаболизм меди в норме
С пищей за сутки в организм поступает приблизительно 2–5 мг меди. В желудочно-кишечном тракте медь транспортируется в эпителий проксимальной части тонкой кишки, где 40–75 % хелатно связывается в клетках специфическим белком металлотионеином и затем экскретируется с фекалиями при десквамации эпителия, а 25–60 % по системе воротной вены при участии специфического переносчика — продукта гена болезни Менкеса поступает в печень, где значительная часть (около 90 %) связывается с синтезируемым печенью транспортным белком — церулоплазмином, циркулирует в сыворотке крови, захватывается избирательно органами, которые в ней нуждаются, и затем выводится преимущественно с желчью, и лишь небольшая часть поступившей меди, минуя печень, попадает в системный кровоток и экскретируется почками. В гепатоцитах медь включается в специфические металлоэнзимы: цитохромоксидазу, моноаминооксидазу, супероксиддисмутазу. Связывание меди с церулоплазмином происходит в аппарате Гольджи при участии продукта гена медьтранспортирующего АТФазного протеина Р-типа, который, как предполагается, принимает активное участие в лизосомальной экскреции меди. С желчью выводится около 80 % поступившей в печень меди, при этом в желчи медь связана с крупномолекулярными белками, что препятствует ее реабсорбции в тонкой кишке и предотвращает энтерогепатическую циркуляцию меди. Таким образом, количества поступившей и выделенной меди становятся практически равными.
Механизмы токсичности меди
Медь, являясь прооксидантом, катализирует образование свободных гидроксильных радикалов и запускает процессы перекисного окисления липидов. Избыточное ее накопление вызывает нарушение функции плазматической мембраны, мембран митохондрий, выхода лизосомальных энзимов в клетку, нарушение функционирования ДНК и белков, снижение содержания антиоксидантов (глутатиона и токоферола) в тканях. В результате перекисного окисления липидов образуется малоновый альдегид, который стимулирует синтез коллагена, что способствует фиброгенезу. Свободная медь, накапливающаяся в тканях, блокирует SH-группы глутатиона и многих ферментов, участвующих в окислительно-восстановительных реакциях, что приводит к энергетическому голоданию. В печени потенциальными мишенями действия оксидантов являются митохондрии. Нарушение дыхательной цепи митохондрий и снижение активности цитохром-С-оксидазы еще больше увеличивает продукцию свободных радикалов. Вследствие недостаточного использования меди она последовательно депонируется в органах и системах. Прежде всего в печени, а затем в центральной нервной системе, в роговице, органах кроветворения, почках, коже, сердце, костно-суставной и эндокринной системах. Механизмами, защищающими гепатоцит от избытка меди, являются ее детоксикация при связывании с глутатионом и металлотионеином, билиарная экскреция с участием лизосом и других медьтранспортирующих систем.
Распространенность
Болезнь распространена повсеместно. Частота гена болезни Вильсона — Коновалова оценивается различно: от 1 на 200 до 1 на 500 человек. Частота заболевания разными авторами также оценивается различно: 1–5 на 30–300 тысяч населения. Имеются данные, что чаще болеют мужчины. Частота выше в регионах, где существуют близкородственные браки (Иран, Йемен, Иордания, Япония, Индия, южные регионы Италии), и у евреев восточноевропейского происхождения. Имеются данные о конкордантности по болезни Вильсона — Коновалова у однояйцевых близнецов. Гетерозиготные носители гена составляют по разным данным 1 на 22–90 человек.
Этиология
Заболевание наследуется по аутосомно-рецессивному типу, опосредуется рецессивным геном, расположенным на длинном плече 13-й хромосомы и экспрессирующимся в печени, почках, плаценте. Его продукт — катион, транспортирующий Р-тип АТФазного протеина (АТР7В), локализуется в цитоплазме и аппарате Гольджи клеток печени и головного мозга. Мутация гена приводит к нарушению структуры транспортного белка, что обусловливает нарушение экскреции меди желчью и синтеза церулоплазмина. В настоящее время описано более 40 различных мутаций этого гена, что объясняет различную степень нарушения транспорта меди и соответственно различие в клинической картине и биохимических данных в семьях больных.
Патогенез
В патогенезе заболевания ведущая роль принадлежит нарушению баланса между поступлением меди и ее экскрецией. Интестинальная абсорбция меди не изменена, но отмечается значительное снижение выведения меди желчью за счет лизосомальной фракции, что приводит к ее накоплению в гепатоцитах. Такая ситуация может быть связана с дефицитом или полным отсутствием продукта гена, который осуществляет транспорт меди в аппарате Гольджи и последующее выделение лизосомами в желчь. При этом нарушается процесс включения меди в апоцерулоплазмин, что приводит к снижению содержания церулоплазмина и увеличению уровня свободной меди в крови, к депонированию ее в органах и тканях, а также к повышенному выделению с мочой.
Церулоплазмин синтезируется исключительно в печени — в цитоплазме гепатоцитов вокруг ядра. При болезни Вильсона — Коновалова прерывается синтез этого транспортного белка. Снижение или отсутствие активности церулоплазмина нарушает поступление достаточных количеств меди к ферментам тканевого дыхания, кроветворным органам. В свою очередь депонированная в печени медь вторично ингибирует синтез церулоплазмина, что приводит к снижению и без того недостаточного его содержания.
Таким образом, при болезни Вильсона — Коновалова имеется, с одной стороны, недостаточное поступление меди в ферментные системы и вследствие этого нарушение биологических процессов, с другой — накопление меди в тканях с последующей интоксикацией этим металлом.
Течение заболевания
В течении заболевания можно выделить три стадии:
I стадия. Медь накапливается в цитозоле печеночных клеток (экстрализосомально); связанная SH-группами протеинов цитозоля она затрудняет секрецию гепатоцитами белков и триглицеридов. Клинические признаки заболевания отсутствуют.
II стадия. Медь перераспределяется из цитозоля в лизосомы гепатоцитов, часть ее поступает в кровь; экскреция меди желчью снижается. Повышение концентрации меди в лизосомах приводит к усилению перекисного окисления липидов и повреждению лизосомальных мембран с последующим выходом кислых гидролаз в цитоплазму гепатоцитов. Наблюдаются некроз гепатоцитов, развитие хронического гепатита и гемолитической анемии.
III стадия. Медь усиленно накапливается в печени, что приводит к фиброзу и циррозу печени, а также в других органах — головном мозге, роговице, дистальных отделах почечных канальцев, что тоже способствует развитию развернутой картины заболевания с характерными клиническими признаками.
Клиническая картина
Основными и зачастую первыми в клинической картине являются симптомы поражения печени, которые отмечаются уже в 5–6-летнем возрасте и раньше.
Острый гепатит развивается примерно у 1/4 пациентов. У больных появляется желтуха, астенический синдром, симптомы общей интоксикации. При биохимическом исследовании крови определяется повышение билирубина, активности аминотрансаминаз. Формируются Кумбс-отрицательная анемия и снижение уровня мочевой кислоты. При морфологическом исследовании биоптата печени определяется отек гепатоцитов, единичные некрозы, умеренная лимфоцитарная инфильтрация. Указанные изменения могут самостоятельно разрешаться, и наступает временное клиническое улучшение, но печеночные функциональные пробы остаются измененными. При дальнейшем прогрессировании процесса происходит хронизация заболевания.
Хронический гепатит — наиболее частый вариант манифестации болезни Вильсона — Коновалова у подростков и молодых пациентов. Характеризуется всеми клиническими и биохимическими признаками данного заболевания: гепатит с высокой степенью активности, желтухой, умеренным уровнем аминотрансфераз, гипергаммаглобулинемией, Кумбс-отрицательной гемолитической анемией, низким уровнем мочевой кислоты, семейным анамнезом. При биопсии печени выявляются баллонная дистрофия, некрозы гепатоцитов, лимфоидная воспалительная инфильтрация, гликогеновая вакуолизация ядер гепатоцитов, перипортальный стеатоз, фиброзирование портальных трактов. При прогрессировании процесса развивается цирроз печени с портальной гипертензией и печеночной недостаточностью.
Фульминантная печеночная недостаточность — редкая и наиболее неблагоприятная в прогностическом отношении форма поражения печени при болезни Вильсона — Коновалова. Часто развивается у подростков и молодых пациентов, практически всегда заканчивается летальным исходом. По клинической и лабораторной картине эта форма сходна с печеночной недостаточностью инфекционной или токсической этиологии, но отличается умеренным повышением уровня аминотрансфераз и низким уровнем щелочной фосфатазы в крови. Характерными признаками печеночной недостаточности при болезни Вильсона — Коновалова является наличие Кумбс-отрицательной анемии вследствие массивного высвобождения меди из печени и повышения содержания сывороточной меди. Фульминантная печеночная недостаточность морфологически проявляется мелкокапельным ожирением, массивными коагуляционными некрозами гепатоцитов, коллапсом стромы, наличием телец Мэллори, гипертрофией клеток Купфера, содержащих большое количество пигмента. Одним из диагностических методов является определение меди в ткани печени (при болезни Вильсона — Коновалова содержание меди свыше 55 мкг/г сухого вещества ткани печени).
Цирроз печени при болезни Вильсона — Коновалова может длительное время протекать бессимптомно или малосимптомно. Его клинические проявления и осложнения, а также данные лабораторных тестов не отличаются от таковых при другой этиологии цирроза. При биопсии печени обнаруживают ложные дольки с фиброзными тяжами вокруг них, гликогеновую вакуолизацию ядер гепатоцитов, отложение темного пигмента в отдельных гепатоцитах (рис. 1). У молодых пациентов с циррозом печени, гемолитической Кумбс-отрицательной анемией, нейропсихической симптоматикой, признаками поражения роговицы и семейным анамнезом по болезни Вильсона — Коновалова обязательно необходимо исключать гепатолентикулярную дистрофию.
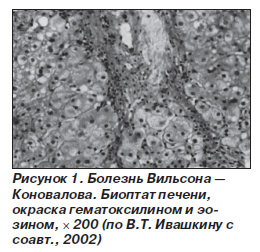 Отдельно выделяют абдоминальную форму Керара, при которой поражение печени преобладает на всем протяжении заболевания и рано осложняется печеночной недостаточностью. Также следует отметить, что у многих пациентов патология печени при болезни Вильсона — Коновалова может долгие годы протекать бессимптомно или иметь стертую клиническую картину, поэтому заболевание чаще диагностируют лишь при появлении неврологической или психической симптоматики, признаков поражения роговицы (наличие колец Кайзера — Флейшера).
Отдельно выделяют абдоминальную форму Керара, при которой поражение печени преобладает на всем протяжении заболевания и рано осложняется печеночной недостаточностью. Также следует отметить, что у многих пациентов патология печени при болезни Вильсона — Коновалова может долгие годы протекать бессимптомно или иметь стертую клиническую картину, поэтому заболевание чаще диагностируют лишь при появлении неврологической или психической симптоматики, признаков поражения роговицы (наличие колец Кайзера — Флейшера).
После того как печень насыщается медью, этот металл накапливается последовательно и в других органах и системах, прежде всего в центральной нервной системе, что приводит к появлению неврологических проявлений, которые чаще всего развиваются во втором и третьем десятилетиях жизни больных. Признаки поражения этой системы имеются у 34 и 10 % пациентов с болезнью Вильсона — Коновалова соответственно. Среди всех симптомов можно выделить флексорно-экстензорный тремор, выраженность которого варьирует от едва заметного дрожания рук до трясения всего тела. Типичным считают «порхающий» тремор пальцев вытянутых рук, усиливающийся при волнении и целенаправленных действиях. У всех больных отмечается различной степени мышечная дистония. Для дрожательно-ригидной формы характерны сочетание тремора и ригидности, гипомимия, гиперсаливация, затрудненная монотонная речь. Акинетико-ригидная форма проявляется выраженной ригидностью в различных группах мышц. На поздних стадиях заболевания появляется характерный гиперкинез по типу «бьющихся крыльев», дизартрия и дисфагия, мозжечковые расстройства, миоклония, макроглоссия. Примерно у 6 % пациентов отмечаются эпилептические припадки, что в 10 раз выше, чем в общей популяции. Ригидность с явными признаками паркинсонизма, сгибательные контрактуры, большие эпилептические припадки и мышечная спастичность встречаются гораздо реже и в основном на поздних стадиях заболевания. Познавательная функция обычно сохраняется, хотя и отмечается снижение интеллекта.
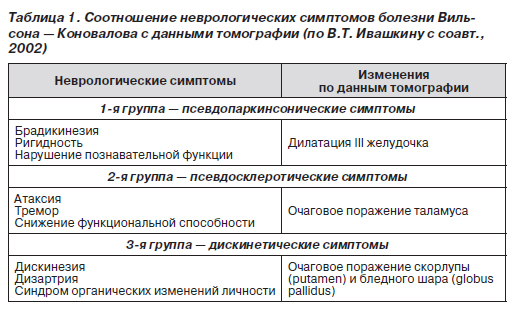
Выделяют три группы неврологических симптомов, которые соотносятся со структурными изменениями по данным магнитно-резонансной томографии (табл. 1).
Психические расстройства развиваются примерно у 1/3 пациентов с болезнью Вильсона — Коновалова и практически у всех больных с неврологической симптоматикой. Отмечаются изменение поведения, снижение работоспособности и способности к обучению у детей, изменения личности, лабильность настроения, повышенная эмоциональность, импульсивное и антисоциальное поведение, депрессия, склонность к суицидальным попыткам, гиперсексуальность.
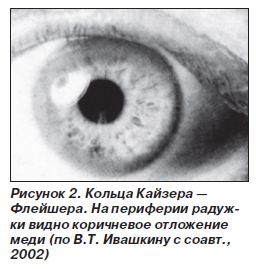
Офтальмологические изменения появляются приблизительно одновременно с неврологической симптоматикой. Характерно появление на роговице колец, описанных независимо друг от друга двумя офтальмологами Кайзером в
Поражение почек при болезни Вильсона — Коновалова проявляется появлением периферических отеков. При исследовании мочи обнаруживается микро- (и гораздо реже макрогематурия), незначительная протеинурия. Повышена концентрация креатинина в сыворотке крови. Вследствие токсического действия меди на почечные тубулярные клетки развиваются проксимальные тубулярные дефекты, вызывающие нарушение гломерулярной фильтрации и проявляющиеся аминоацидурией, глюкозурией, гиперфосфатурией, гиперкальциурией, что может способствовать развитию нефролитиаза.
Гематологические изменения. Течение болезни Вильсона — Коновалова часто осложняет острый внутрисосудистый гемолиз. Его развитие связывают с попаданием большого количества меди в кровь, которая оказывает токсическое действие на мембраны эритроцитов и гемоглобин. Гемолиз обычно преходящий, может предшествовать печеночной манифестации заболевания (у 15 % пациентов) или наблюдаться одновременно с острой печеночной недостаточностью.
Костно-суставной синдром при гепатолентикулярной дистрофии проявляется остеопенией, обусловленной остеопорозом или остеомаляцией. Характерно поражение крупных суставов (бедренных, коленных, суставов запястья и позвоночника), что вероятнее всего связано с гиперкальциурией и гиперфосфатурией. При рентген-исследовании определяются дегенеративные изменения: остеофиты, склероз, подхрящевые псевдокисты, фрагментация кости.
Из редких, но характерных поражений других органов и систем следует отметить гипертрофию миокарда левого или обоих желудочков, аритмии, ортостатическую гипотензию, холелитиаз, нарушения полового созревания, изменения гуморального и клеточно-опосредованного иммунитета, гиперпигментацию кожи, появление голубых лунок у ногтевого ложа.
Диагностика
Исключение болезни Вильсона — Коновалова необходимо проводить у пациентов в возрасте от 3 до 40 лет, имеющих увеличение уровня аминотрансфераз, признаки поражения печени, неврологические и психические симптомы неуточненной этиологии, особенно в сочетании с признаками поражения печени, при наличии колец Кайзера — Флейшера и приобретенной гемолитической анемии, а также у пациентов с семейным анамнезом по болезни Вильсона — Коновалова.
Важное место в диагностике этого заболевания отводится типичной клинической картине, которая была описана выше (рис. 3). Характерными признаками при лабораторном обследовании являются анемия, выраженная желтуха, относительно низкая активность аминотрансфераз, снижение или отсутствие активности церулоплазмина в сыворотке крови (менее 20 мг/дл), повышение в сыворотке крови уровня меди, не связанной с церулоплазмином (свыше 300 мкг/л), повышение экскреции меди с мочой (более 100 мкг/сут). Алгоритм ведения больных с подозрением на болезнь Вильсона — Коновалова представлен на рис. 4.

При обычном морфологическом исследовании печени специфические изменения не выявляются. Необходимо гистохимическое исследование пунктата, при котором срезы окрашиваются рубеановой кислотой или роданином, и в них качественно определяют повышенное содержание меди в ткани печени (рис. 5). Одним из дополнительных тестов в диагностике заболевания служит количественное определение меди в печени (у пациентов с болезнью Вильсона — Коновалова содержание от 250 до 3000 мкг/г).

В сложных диагностических случаях изучают поглощение печенью радиоактивной меди и проводят генетические исследования.
Лечение
 Прежде всего назначают диету, предусматривающую уменьшение употребления продуктов, содержащих медь (необработанная пшеница, бобовые, шоколад, печень, почки, моллюски и др.).
Прежде всего назначают диету, предусматривающую уменьшение употребления продуктов, содержащих медь (необработанная пшеница, бобовые, шоколад, печень, почки, моллюски и др.).
Лекарственная терапия направлена на выведение избытка меди из организма для предотвращения ее токсического влияния. С момента установления диагноза или выявления гомозиготного носительства дефектного гена болезни Вильсона — Коновалова пациенты должны получать патогенетическую терапию на протяжении всей жизни.
Британский антилюизит. Увеличивает выведение меди с мочой, снижая ее содержание в организме. Вводится по 1,25–2,5 мг/кг 2 раза в день внутримышечно в течение 10–20 дней, перерыв между курсами 20 дней. В настоящее время применяют редко из-за выраженной болезненности в месте инъекции, в основном у пациентов с прогрессивным ухудшением состояния и развитием неврологической симптоматики.
Унитиол. Вводится по 5–10 мл ежедневно или через день внутримышечно, на курс 25–30 инъекций; перерыв 2–3 месяца.
D-пеницилламин. Предполагается, что он увеличивает выведение меди с мочой, так как образует такие соединения с медью, которые легко фильтруются через почечные клубочки, индуцирует синтез металлотионеинов и перевод внутриклеточной меди в неактивное состояние. Кроме того, этот препарат ингибирует синтез коллагена, увеличивает внутриклеточный уровень глутатиона, уменьшает воспалительную реакцию. Оптимальной дозой препарата считается 0,9–2,0 г/сут. Начальная доза составляет 250–500 мг/сут, разделенная на 4 приема. Затем дозу препарата постепенно повышают каждые 7 дней на 250 мг, пока экскреция меди с мочой не повысится до 2000–5000 мкг/сут. После достижения клинического улучшения, которое наступает через несколько месяцев после начала лечения, переходят на поддерживающую терапию (0,75–1,25 г/сут). Следует помнить, что при лечении D-пеницилламином возможно развитие побочных эффектов. К ранним побочным эффектам, развивающимся во время начальной фазы лечения, относят ухудшение неврологической симптоматики (как правило, в течение первого месяца терапии), развитие реакции гиперчувствительности (лихорадка, кожный зуд, сыпь и лимфаденопатия), в редких случаях значительное угнетение кроветворения с развитием лейко- и тромбоцитопении. Поздние эффекты развиваются на фоне поддерживающей терапии — кожные проявления, синдромы, сходные с аутоиммунными заболеваниями. Во всех случаях побочных эффектов дозу D-пеницилламина необходимо корригировать: от временного снижения с последующим возвратом к первоначальной дозе до полной отмены препарата и замены его другими медьхелатирующими средствами (в зависимости от развившегося осложнения). Лечение проводят пожизненно.
Триентин. Используется как альтернативный D-пеницилламину медь-хелатирующий агент. Препарат принимается натощак, его доза составляет 1–2 г/сут, разделенная на 3 приема. Одним из требующих коррекции дозы побочных эффектов является сидеробластная анемия.
Цинк. Сульфат цинка менее токсичный, чем D-пеницилламин. Механизм его действия заключается в торможении абсорбции меди в кишечнике. Принимается до еды по 200 мг 3 раза в день. Из наиболее частых побочных эффектов следует отметить расстройства со стороны желудочно-кишечного тракта.
Тетратиомолибдат. Образует в желудочно-кишечном тракте и в сыворотке крови комплексы с медью, тем самым препятствуя ее абсорбции и проникновению в ткани. Потенциально более эффективен, чем D-пеницилламин. Суточная доза препарата составляет 120–200 мг. В качестве побочного эффекта возможно угнетение кроветворения.
Трансплантация печени. Показаниями к ортотопической трансплантации печени при болезни Вильсона — Коновалова являются фульминантная печеночная недостаточность с гемолизом и гиперурикемией и прогрессирующая печеночная недостаточность на фоне хронического гепатита и цирроза печени как проявления основного заболевания, которые не корригируется медикаментозно.
Также в последние годы обсуждается возможность генной терапии болезни Вильсона — Коновалова. Однако замещение в печени дефектного гена заболевания нормальным представляет собой значительные трудности, и этот вопрос до сих пор дискутируется.
Прекращение патогенетического лечения может привести к развитию необратимых изменений в органах и тканях с утратой их функций, к летальному исходу.
Прогноз
Течение заболевания прогрессирующее, приводящее к инвалидности. При своевременном начале лечения (на ранних стадиях болезни, до развития структурных и функциональных изменений в органах и тканях) прогноз значительно улучшается.