
Газета «Новости медицины и фармации» Гастроэнтерология (304) 2009 (тематический номер)
Вернуться к номеру
Наследственный панкреатит
Авторы: Т.Н. Христич, Т.Б. Кендзерская, В.П. Пишак, Н.Б. Губергриц, Буковинский государственный медицинский университет, г. Черновцы; Донецкий национальный медицинский университет им. М. Горького
Версия для печати
Генетика — наука, объясняющая,
Среди множества этиологических факторов, вызывающих патологию поджелудочной железы (ПЖ), пожалуй, наименее изученным является наследственность. С генетической предрасположенностью связывают целый ряд заболеваний ПЖ, но основными из генетически обусловленных неэндокринных заболеваний органа считают муковисцидоз, наследственный панкреатит (НП) и наследственный рак ПЖ. Не случайно на логотипе Midwest Multicentre Pancreatic Study Group, созданной для всестороннего изучения наследственной патологии ПЖ (эпидемиологии, патогенеза, клинических проявлений, прогноза и т.д.), указаны именно эти три заболевания (рис. 1).
 Кроме них наследственными являются также: синдром Швахмана, синдром Йохансона — Близзарда, врожденная сидеробластная анемия с экзокринной недостаточностью ПЖ, синдром Кларка — Хэдвилда, синдром Андерсена, изолированная недостаточность отдельных панкреатических ферментов (липазы, колипазы, амилазы, трипсиногена), синдром недостаточности энтерокиназы, макроамилаземия, дефицит a1-антитрипсина и др. [10]. Безусловно, генетически обусловленными являются также аномалии развития ПЖ: pancreas divisum, pancreas annulare, pancreas aberrans, врожденные кисты ПЖ и др. [3, 4, 18].
Кроме них наследственными являются также: синдром Швахмана, синдром Йохансона — Близзарда, врожденная сидеробластная анемия с экзокринной недостаточностью ПЖ, синдром Кларка — Хэдвилда, синдром Андерсена, изолированная недостаточность отдельных панкреатических ферментов (липазы, колипазы, амилазы, трипсиногена), синдром недостаточности энтерокиназы, макроамилаземия, дефицит a1-антитрипсина и др. [10]. Безусловно, генетически обусловленными являются также аномалии развития ПЖ: pancreas divisum, pancreas annulare, pancreas aberrans, врожденные кисты ПЖ и др. [3, 4, 18].
Наша статья в связи с лимитом объема не может охватить все наследственные заболевания ПЖ. Остановимся подробно лишь на НП. Впервые НП был описан M.W. Comfort и A.Е. Steinberg в
Однако до точного объяснения патогенеза НП существовали предположения о связи заболевания с наследственной патологией протоков. Согласно этой гипотезе, первичными являются аномалии протоковой системы ПЖ, а развитие панкреатита — вторичным (из-за нарушения оттока панкреатического секрета). Эта гипотеза была основана на том, что при НП часто выявляются изменения протоковой системы ПЖ. Выдвигалась также теория дефицита антиоксидантов в ткани ПЖ, согласно которой основное значение в патогенезе придавалось оксидантному стрессу [3, 4, 10].
В
Позже были выявлены мутации гена серинпротеазного ингибитора Казаля типа I (SPINK I) у пациентов с идиопатическим хроническим панкреатитом, доказана связь около половины случаев тропического панкреатита и части случаев идиопатического хронического панкреатита с мутацией SPINK I (N291) [8, 11, 15, 16].
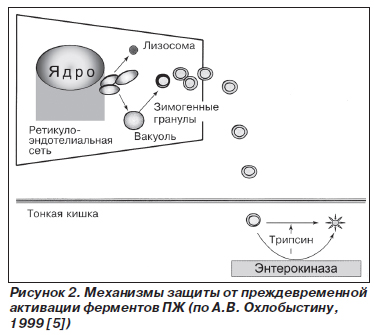
Учитывая, что ПЖ синтезирует множество пищеварительных ферментов, существует целый ряд дублирующих друг друга механизмов, препятствующих аутолизу ткани органа (рис. 2) [4, 20]:
 — локализация ферментов в зимогенных гранулах ацинарных клеток, предотвращающих выход ферментов в цитоплазму;
— локализация ферментов в зимогенных гранулах ацинарных клеток, предотвращающих выход ферментов в цитоплазму;
В принципе, мутация любого из генов, кодирующих механизмы самозащиты ПЖ, может привести при наличии внешних провоцирующих факторов к развитию панкреатита.
Проанализируем подробнее патогенез НП, ассоциированного с мутациями R122H и N291. Молекула трипсина состоит из двух субъединиц, соединенных полипептидной цепью (рис. 3). В положении 117 этой цепи находится аргинин. Между двумя субъединицами трипсина располагается его активный центр, который способен распознавать аргинин и лизин и осуществлять в месте соединения этих аминокислот лизис полипептидной цепи. Именно таким образом трипсин, мезотрипсин и энзим Y инактивируют 80 % интрапанкреатических трипсиногена и трипсина [4, 21]. Остальные 20 % инактивации интрапанкреатических протеаз обеспечиваются SPINK I (рис. 4).
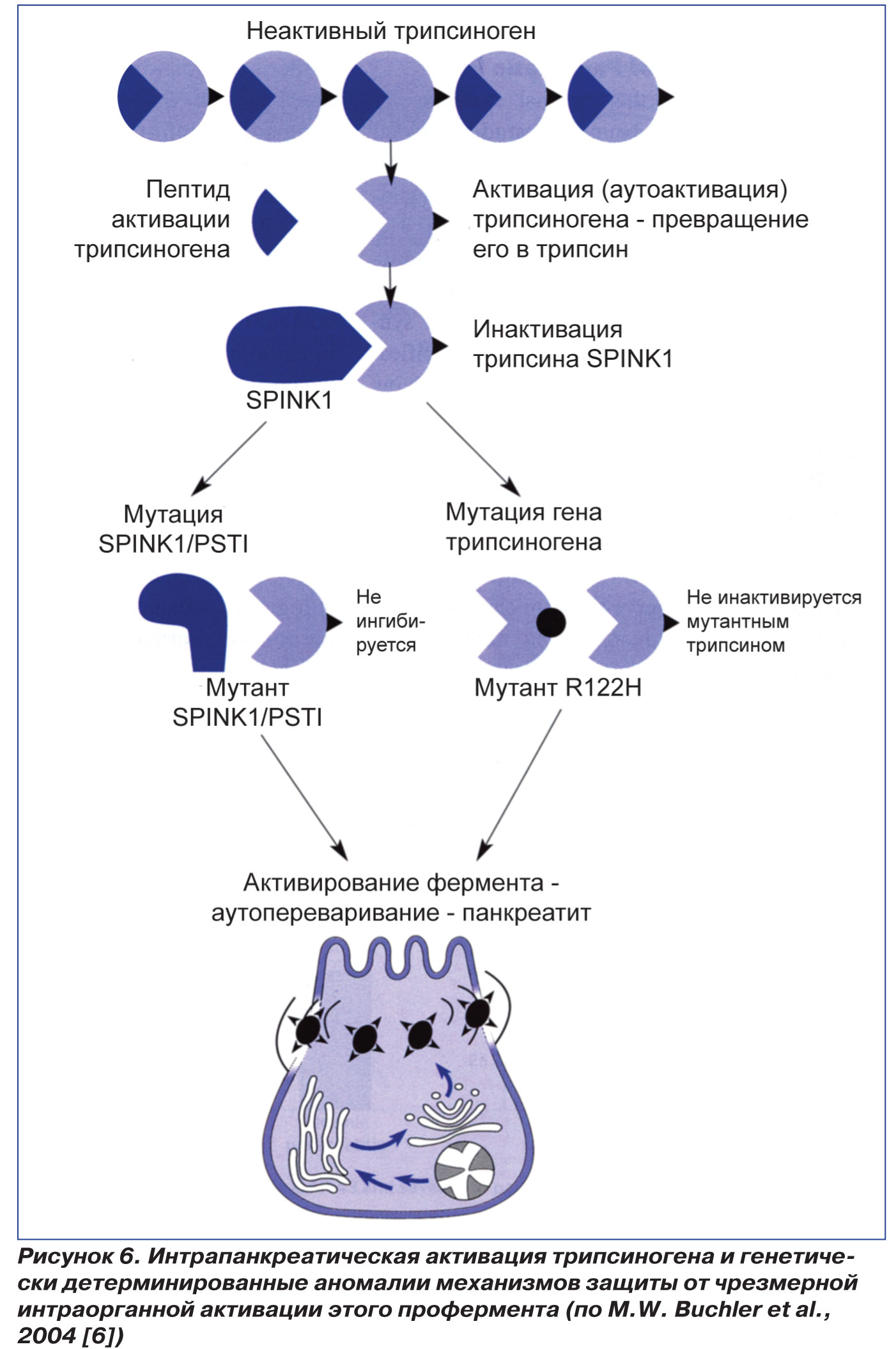
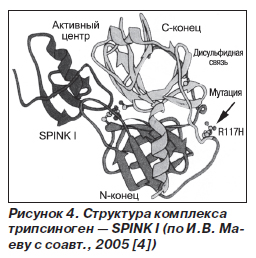 Этот ингибитор представляет собой специфический субстрат для трипсина. SPINK I необратимо связывает серин трипсина с лизином своего активного центра. Важно, что SPINK I синтезируется в 20 раз меньше, чем количество трипсиногена, продуцируемого ПЖ. В связи с этим SPINK I может полностью ингибировать трипсин в ткани органа только тогда, когда уровень активности трипсина низкий. В этих случаях SPINK I предотвращает последующую аутоактивацию трипсиногена и блокирует каскад активации панкреатических ферментов и аутолиза ПЖ (рис. 5). При интенсивной активации трипсиногена SPINK I не в состоянии его инактивировать. В этом случае трипсин и другие трипсиноподобные ферменты, как было сказано выше, лизируют полипептидную цепь, объединяющие 2 субъединицы трипсина, в положении 117, т.е. в месте соединения аргинина и лизина. При мутации катионного трипсиногена R122H аргинин заменяется на гистидин, поэтому трипсин не способен лизировать молекулы трипсиногена и трипсина. Мощности SPINK I в этих случаях не хватает для блокирования аутоактивации трипсиногена, продолжается каскад активации панкреатических ферментов и аутолиз ПЖ [4, 10]. При мутации SPINK I (N291) снижается степень инактивации трипсина, и при воздействии мощного провоцирующего фактора (алкоголь) также развивается НП (рис. 6).
Этот ингибитор представляет собой специфический субстрат для трипсина. SPINK I необратимо связывает серин трипсина с лизином своего активного центра. Важно, что SPINK I синтезируется в 20 раз меньше, чем количество трипсиногена, продуцируемого ПЖ. В связи с этим SPINK I может полностью ингибировать трипсин в ткани органа только тогда, когда уровень активности трипсина низкий. В этих случаях SPINK I предотвращает последующую аутоактивацию трипсиногена и блокирует каскад активации панкреатических ферментов и аутолиза ПЖ (рис. 5). При интенсивной активации трипсиногена SPINK I не в состоянии его инактивировать. В этом случае трипсин и другие трипсиноподобные ферменты, как было сказано выше, лизируют полипептидную цепь, объединяющие 2 субъединицы трипсина, в положении 117, т.е. в месте соединения аргинина и лизина. При мутации катионного трипсиногена R122H аргинин заменяется на гистидин, поэтому трипсин не способен лизировать молекулы трипсиногена и трипсина. Мощности SPINK I в этих случаях не хватает для блокирования аутоактивации трипсиногена, продолжается каскад активации панкреатических ферментов и аутолиз ПЖ [4, 10]. При мутации SPINK I (N291) снижается степень инактивации трипсина, и при воздействии мощного провоцирующего фактора (алкоголь) также развивается НП (рис. 6).

НП подразделяется на:
1) классический (аутосомно-доми-нантный, с пенетрантностью 80 %; ген катионного трипсиногена PRSS1, R122H, N291);
2) идиопатический (PRSS1 — A16V, D22G, K23R).
Отдельно выделяют особые формы НП: панкреатит с гипераминоацидурией (выделение с мочой аминокислот в результате генетического тубулярного дефекта); геморрагический панкреатит с увеличением выделения электролитов с потом; панкреатит, обусловленный нарушением обмена кальция.
В связи с новыми представлениями о роли генных мутаций в патогенезе хронического панкреатита можно предположить, что две ведущие формы идиопатического панкреатита — с ранним и поздним началом — обусловлены разными генетическими механизмами. Более того, вариант с поздним началом может быть модифицирован факторами среды, такими, например, как алкоголь. В соответствии с этой гипотезой возможно, что среди алкоголиков хронический панкреатит развивается только у лиц с генетической предрасположенностью к этой патологии. Это подтверждается тем, что алкогольный хронический панкреатит диагностируется лишь у небольшой части людей, злоупотребляющих спиртными напитками. С этим согласуется тот факт, что клиническая манифестация доказанного НП появляется обычно в возрасте 3–10 лет и имеет второй пик в возрасте 20–25 лет, когда большинство пациентов начинают принимать алкоголь [4, 10].
В развитии НП доказано значение и других мутаций. В частности, мутации CFTR — гена трансмембранного регулятора кистозного фиброза. Эта мутация и мутация SPINK I наследуются по аутосомно-рецессивному типу. При наличии у пациента мутации CFTR развивается муковисцидоз или генетически детерминированный гипоферментный панкреатит. Такой вариант НП возможен также при недостаточности продукции и нарушении активации трипсиногена [10].
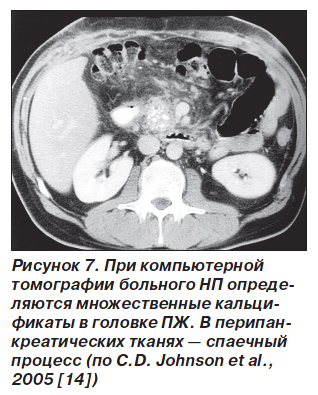
Идентификация НП традиционными методами невозможна, поскольку специфических морфологических и биохимических маркеров не существует [2]. При обследовании каждого пациента необходимо проводить тщательный сбор анамнеза, учитывать наследственность, никотиново-алкогольную зависимость, потерю массы тела. Хотя НП характеризуется ранним началом (у 80 % больных приходится на возраст до 20 лет), но практически всегда — поздней диагностикой, образованием кальцификатов ПЖ округлой формы (50 % случаев) (рис. 7), псевдокист.
 Начальные его проявления сходны с клиникой острого панкреатита. Типичны повторные эпизоды болевого абдоминального и диспептического синдромов с постепенно увеличивающейся частотой и выраженностью рецидивов, нарастанием степени функциональной недостаточности ПЖ (у 15–20 % больных выявляется выраженная стеаторея), что приводит к развитию хронического панкреатита. Для НП характерно увеличение длительности ремиссий с течением времени. В поздних стадиях заболевания развиваются сахарный диабет (присоединяется через 8–10 лет у 20 % больных), тромбозы крупных вен (воротной, селезеночной, нижней полой), геморрагии [4, 17]. Панкреатические атаки при НП не отличаются от других форм хронического панкреатита. Однако последние исследования доказали, что нельзя считать злоупотребление алкоголем или наличие холелитиаза факторами, исключающими НП. Это говорит о том, что изменение трипсина является лишь фоном. Мутация приводит не к усиленной активации трипсина, а лишь нарушает один из защитных механизмов ацинарных клеток. Для возникновения заболевания, появления клинических симптомов необходимы инициирующие факторы, которые вызвали бы эту активацию. Поэтому для верификации диагноза НП необходимо выявление мутаций трипсиногена с помощью полимеразной цепной реакции.
Начальные его проявления сходны с клиникой острого панкреатита. Типичны повторные эпизоды болевого абдоминального и диспептического синдромов с постепенно увеличивающейся частотой и выраженностью рецидивов, нарастанием степени функциональной недостаточности ПЖ (у 15–20 % больных выявляется выраженная стеаторея), что приводит к развитию хронического панкреатита. Для НП характерно увеличение длительности ремиссий с течением времени. В поздних стадиях заболевания развиваются сахарный диабет (присоединяется через 8–10 лет у 20 % больных), тромбозы крупных вен (воротной, селезеночной, нижней полой), геморрагии [4, 17]. Панкреатические атаки при НП не отличаются от других форм хронического панкреатита. Однако последние исследования доказали, что нельзя считать злоупотребление алкоголем или наличие холелитиаза факторами, исключающими НП. Это говорит о том, что изменение трипсина является лишь фоном. Мутация приводит не к усиленной активации трипсина, а лишь нарушает один из защитных механизмов ацинарных клеток. Для возникновения заболевания, появления клинических симптомов необходимы инициирующие факторы, которые вызвали бы эту активацию. Поэтому для верификации диагноза НП необходимо выявление мутаций трипсиногена с помощью полимеразной цепной реакции.
Генетическое тестирование при подозрении на НП позволяет [9]:
Другие методы диагностики НП являются традиционными для верификации хронического панкреатита вообще.
Одна из основных опасностей НП — резкое повышение риска развития рака ПЖ. Так, эпидемиология НП изучалась в двух независимых исследованиях в США и Европе, которые выявили повышение риска развития рака ПЖ у больных НП в 50–70 раз. Эти исследования проводились на основе баз данных Международной группы по изучению НП (International Hereditary Pancreatitis Study Group) и Европейского регистра НП и рака ПЖ (EUROPAC — European Registry of Hereditary Pancreatitis and Pancreatic Cancer). Риск рака ПЖ повышается начиная с 40 лет и достигает 40–70 % в возрасте 70 лет (особенно, если НП прослеживается по мужской линии). Чаще всего рак развивается в семьях с мутациями R122H, N291 или же без мутаций, но с явным фенотипом НП [10].
В то же время при генетических исследованиях больных НП изменений онкогенов или генов-супрессоров опухолевого роста не обнаружено [22]. Это позволяет предположить, что причина частоты возникновения рака ПЖ при НП заключается в более высоком уровне активности воспалительного процесса и большей его длительности. Это лишний довод в пользу того, что само хроническое воспаление является предрасполагающим фактором развития рака ПЖ. Вероятно, имеет значение длительность воспалительного процесса, отсутствие этиотропной терапии при НП. В других исследованиях, напротив, показано, что при раке ПЖ, развившемся вследствие НП, мутации онкогена K-ras встречаются как в клетках опухоли, так и в прилежащих к ней тканях, а также при НП в зонах гиперплазии эпителия протоков [1, 12]. Мутации онкогенов можно выявить при биопсии ПЖ или при исследовании ее сока (обычно для его получения используют секретиновый или секретин-панкреозиминовый тесты). Патогенез развития рака ПЖ при НП представлен на рис. 8.

Большая часть опухолей исходят из протокового эпителия ПЖ. В настоящее время нет данных, подтверждающих различие патогенеза рака ПЖ при НП и при банальном хроническом панкреатите, а также при исходно интактной ПЖ [10].
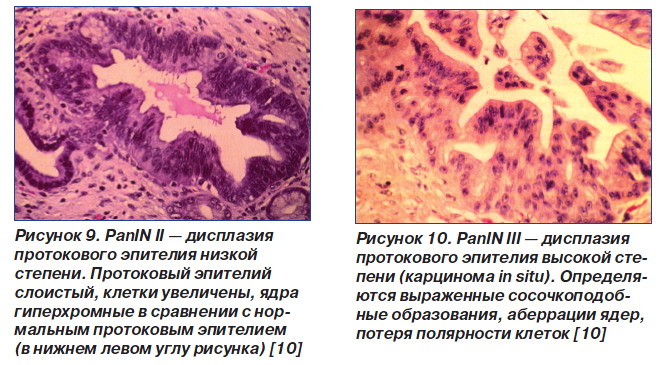
Если у больного НП развились клинические признаки, подозрительные в отношении рака ПЖ, то необходимо провести эндоскопическую ретроградную холангиопанкреатографию (ЭРХПГ) с биопсией клеток панкреатического протока [7]. Доказано, что предраковым состоянием в отношении аденокарциномы ПЖ является панкреатическая дисплазия (панкреатическая внутрипротоковая неоплазия — PanIN). Выделяют 3 степени PanIN [10]:
— PanIN II — дисплазия низкой степени (рис. 9);
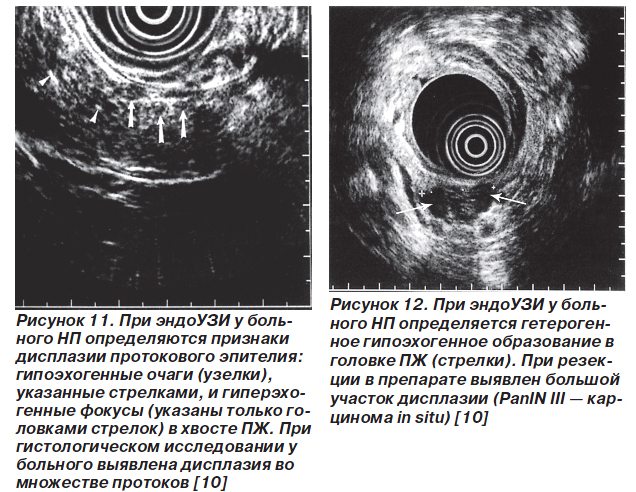

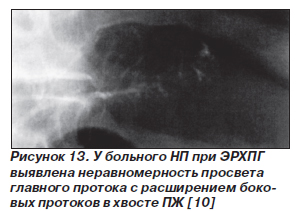 Для скрининга дисплазии протокового эпителия и карциномы in situ на фоне НП применяют эндоскопическую сонографию (эндоУЗИ), ЭРХПГ, спиральную компьютерную томографию, определение уровней карбоантигена 19-9 и карциэмбрионального антигена в крови. Однако достаточно информативными являются только эндоУЗИ и ЭРХПГ. Признаки панкреатической дисплазии при эндоУЗИ — гетерогенность паренхимы, наличие гиперэхогенных фокусов и/или гипоэхогенных узелков (рис. 11, 12). По результатам ЭРХПГ признаком, ассоциированным с дисплазией, считают неравномерное расширение протоков, иногда с мешковидными расширениями (рис. 13). Симптомы дисплазии протокового эпителия ПЖ, которые выявляются при эндоУЗИ и ЭРХПГ, представлены в табл. 1.
Для скрининга дисплазии протокового эпителия и карциномы in situ на фоне НП применяют эндоскопическую сонографию (эндоУЗИ), ЭРХПГ, спиральную компьютерную томографию, определение уровней карбоантигена 19-9 и карциэмбрионального антигена в крови. Однако достаточно информативными являются только эндоУЗИ и ЭРХПГ. Признаки панкреатической дисплазии при эндоУЗИ — гетерогенность паренхимы, наличие гиперэхогенных фокусов и/или гипоэхогенных узелков (рис. 11, 12). По результатам ЭРХПГ признаком, ассоциированным с дисплазией, считают неравномерное расширение протоков, иногда с мешковидными расширениями (рис. 13). Симптомы дисплазии протокового эпителия ПЖ, которые выявляются при эндоУЗИ и ЭРХПГ, представлены в табл. 1.
В апреле
До сих пор не существует каких-либо эффективных схем скрининга для обследования пациентов из группы риска по панкреатической аденокарциноме (в частности, пациентов с НП). Это объясняется следующими причинами:
На том же симпозиуме в Милане были приведены следующие данные [10]. Ежегодный скрининг 250 больных НП в возрасте 40–50 лет в течение 5 лет с использованием спиральной компьютерной томографии, ЭРХПГ, эндоУЗИ, с определением онкомаркеров в сыворотке крови и панкреатическом соке стоил в среднем 362 857 долларов на один установленный диагноз рака ПЖ. Тогда как ежегодное обследование подобной группы пациентов с использованием эндоУЗИ и забором крови и панкреатического сока для хранения (без проведения исследований) обошлось в 69 943 доллара на один диагноз рака ПЖ. Несмотря на неоптимистические выводы, Консенсус, принятый в Милане, утверждает, что больные НП старше 40 лет обязательно должны проходить ежегодный скрининг в отношении рака ПЖ. В настоящее время наиболее информативным является эндоУЗИ, хотя окончательный вывод формулировать еще рано, так как продолжаются исследования по усовершенствованию диагностических методик.
Специфическое лечение НП не разработано. Учитывая быстрое прогрессирование функциональной недостаточности ПЖ, показано назначение заместительной ферментной терапии уже на ранних стадиях заболевания. Безусловно, препаратом выбора в этом отношении является креон, имеющий существенные преимущества перед другими ферментными препаратами (минимикросферическая форма выпуска, высокая активность ферментов, особенно липазы, кислотоустойчивая оболочка минимикросфер, наличие других липолитических ферментов, оптимальное соотношение колипаза/липаза и др.). Как правило, больные нуждаются в лечении креоном 25 000, так как при использовании креона 10 000 для компенсации функциональной недостаточности ПЖ требуется прием большего количества капсул.
В ряде случаев необходимо назначение ненаркотических анальгетиков в больших дозах, в том числе детям [2]. В единичных работах показано, что прием больших доз антиоксидантов способствует снижению потребности в анальгетиках. Так, доказана эффективность антиоксидантного витаминно-минерального комплекса (сульфаденозил-метионин — 800 мг/сут, витамин С — 180 мг/сут, витамин Е — 30 мг/сут, витамин А — 2,4 мг/сут, селен — 75 мкг/сут). Введение в терапию НП этого комплекса способствовало снижению интенсивности болевого синдрома и потребности в анальгетиках [4, 22].
Подходы к ведению больных НП с аденокарциномой ПЖ схожи с таковыми при опухоли отличного от НП генеза. В Консенсусе высказывается мнение, что у пациентов с НП и аденокарциномой ПЖ нужно проводить панкреатэктомию. Панкреатэктомия является также профилактической мерой при выявлении протоковой дисплазии, обнаруженной с помощью биопсии панкреатического протока, особенно у пациентов старше 30 лет [12]. Хотя результаты таких мероприятий и не верифицированы ни одним из проспективных исследований, логическое рассуждение доказывает необходимость удаления всего органа, так как при НП в ткани ПЖ процессы регенерации выражены значительно и способствуют повышенной частоте малигнизации.
Мы находимся только у истоков изучения НП во всех его аспектах. НП — это одна из загадочных сторон панкреатологии, которую нам предстоит познавать. Все же не стоит во всех случаях панкреатитов с неясной для врача этиологией считать их наследственными. Для такого суждения необходимы только объективные результаты генетического тестирования, которое, к сожалению, в настоящее время недоступно для практических врачей Украины. В отсутствие таких результатов, опираясь только на эмпирические суждения, легко сделать спекулятивное заключение о НП.
1. Кубышкин В.А., Вишневский В.А. Рак поджелудочной железы. — М.: Медпрактика-М, 2003. — 386 с.
2. Кучерявый Ю.А., Демочко Е.И. Наследственный панкреатит с аутосомно-доминантным типом наследования // Врач. — 2004. — № 8. — С. 8-11.
3. Маев И.В. Наследственные болезни поджелудочной железы // Клин. перспективы гастроэнтерол., гепатол. — 2002. — № 4. — С. 20-27.
4. Маев И.В., Казюлин А.Н., Кучерявый Ю.А. Хронический панкреатит. — М.: Медицина, 2005. — 504 с.
5. Охлобыстин А.В. Новые данные о патогенезе наследственного панкреатита // Рос. журн. гастроэнтерол., гепатол., колопроктол. — 1999. — № 4. — С. 18-23.
6. Büchler M.W.,Uhl W., Malfertheiner P., Sarr M.G. Diseases of the pancreas. —
7. Charnley R.M. Hereditary pancreatitis // World J. Gastroenterol. — 2003. — Vol. 9, № 1. — P. 1-4.
8. Drenth J.P.H., te Morsche R., Jan-sen J.B.M.J. Mutations in serine protease inhibitor Kazal type I are strongly associated with chronic pancreatitis // Gut. — 2002. — Vol. 50. — P. 687-692.
9. Etemad B., Whitcomb D.C. Chronic Pancreatitis: Diagnosis, Classification, and New Genetic Developments // Gastroenterology. — 2001. — Vol. 120. — P. 682-707.
10. Genetic Disorders of the Exocrine Pancreas: An Overview and Update / Ed. by P. Durie, M.M. Lerch, A.B. Lowenfels et al. — Basel et al.: Karger, 2002. — 155 p.
11. Gaia E., Salacone P., Gallo M. et al. Germline mutations in CFTR and PSTI genes in chronic pancreatitis patients // Dig. Dis. Sci. — 2002. — Vol. 47, № 11. — P. 2416-2421.
12. Exocrine pancreas cancer: The European Pancreatic Cancer-Research Cooperative (EPC-RC) / Ed. by T.M. Gress, J.P. Neoptolemos, N.R. Lemoine, F.X. Real. — 2005. — 531 p.
13. Whitcomb D.C., Gorry M.C., Preston R.A. et al. Hereditary pancreatitis is caused by a mutation of cathionic trypsinogen gene // Natural Genet. — 1996. — Vol. 14. — P. 141-145.
14. Johnson C.D., Schmit J.D. Mayo Clinic Gastoreintestinal Imaging Review. — Rochester: Mayo Clinic Scientific Press, 2005. — 737 p.
15. Mössner J. Surveillance and diagnostics of chronic pancreatitis // Disease pro-gression and Disease Prevention in Hepatology and Gastroenterology (Falk Symposium № 50). — Berlin, 2005. — P. 35-36.
16. Witt H., Luck W., Hennies H.C. et al. Mutations in the gene encoding the serine protease inhibitor, Kazal type I are associated with chronic pancreatitis // Nat. Genet. — 2000. — Vol. 25. — P. 213-216.
17. Paolini O., Hastier P., Buckley et al. The natural history of hereditary chronic pancreatitis: a study of 12 cases compared to chronic alcoholic pancreatitis // Pancreas. — 1998. — Vol. 17. — P. 226-271.
18. The Pancreas / Ed. by H.G. Beger et al. — Oxford et al.: Blackwell Science Ltd., 1998. — Vol. 1. — 885 p.
19. Pfutzer R.H., Barmada M.M., Brunskil A.P. SPINK 1/PST 1 polymorphisms act as disease modifiers in familial and idiopathic of chronic pancreatitis // Gastroentero-logy. — 2000. — Vol. 119. — P. 615-623.
20. Sahin-Toht M., Toht M. Gain-of-function mutations associated with hereditary pancreatitis enhance autoactivation of human cathionic trypsinogen // Biochem. Biophys. Res. Commun. — 2000. — Vol. 279, № 2. — P. 286-289.
21. Sahin-Toht M., Toht M. High-affinity Ca2+ binding inhibits autoactivation of rat trypsinogen // Biochem. Biophys. Res. Commun. — 2000. — Vol. 275, № 2. — P. 668-671.
22. Uomo G., Talamini G., Rabitti G. Antioxidant treatment in hereditary pancreatitis. A pilot study of three young patients // Dig. Liver Dis. — 2001. — Vol. 33, № 1. — P. 58-62.
23. Whitcomb D.C. Hereditary pancreatitis: a model for understanding the genetic basis of acute and chronic pancreatitis // Pancreatology. — 2001. — Vol. 1. — Р. 565-570.