
Газета «Новости медицины и фармации» Антимикробная и противовирусная терапия (310) 2010 (тематический номер)
Вернуться к номеру
Фармакокинетика цефалоспоринов в детском возрасте
Авторы: А.П. Волосовец, С.П. Кривопустов, Национальный медицинский университет им. А.А. Богомольца, г. Киев
Версия для печати
Возраст ребенка определяет особенности абсорбции, распределения, метаболизма, экскреции всех лекарственных препаратов, в том числе антибиотиков, что необходимо учитывать при назначении антибактериальной терапии. Наиболее важными факторами для фармакокинетики антибиотиков являются характер и интенсивность их абсорбции, связанные с функциональными характеристиками желудочно-кишечного тракта ребенка при приеме препарата внутрь, особенности гемодинамики и метаболизма — при парентеральном введении; активность ферментных систем ребенка; объем экстрацеллюлярной жидкости; концентрация белка в плазме крови; степень функциональной зрелости экскреторных органов.
Желудочно-кишечный тракт ребенка, особенно раннего возраста, характеризуется относительно низкой кислотностью желудочного сока с высоким рН. Существенное влияние на желудочно-кишечную абсорбцию антибиотиков оказывает большее, чем у взрослых, соотношение протяженности кишечника и массы тела; чем меньше ребенок, тем это различие более выражено. У детей первых месяцев жизни и новорожденных возможности для абсорбции препаратов значительно выше также за счет большего времени транзита содержимого кишечника и нерегулярной перистальтики.
Немаловажную роль играет более высокая активность фермента двенадцатиперстной кишки бета-глюкуронидазы, которая обусловливает деконъюгацию антибактериальных препаратов, выводимых через гепатобилиарную систему, с последующей реабсорбцией. Раннее детство — это также период становления кишечного биоценоза с постепенным нарастанием нормальной микрофлоры и вытеснением транзиторных условно-патогенных видов микроорганизмов, назначение антибиотиков в этот период также может нарушить процессы становления нормального биоценоза.
Существенное влияние на биотрансформацию лекарственных препаратов оказывает активность ферментных систем новорожденного. Большую роль играют активность глюкуронилтрансферазы (ГТФ) печени, участвующей в конъюгации ряда антибиотиков, и уровень тубулярной экскреции образуемых конъюгатов. До 7-х суток жизни уровень ГТФ снижен, а уровень экскреции конъюгатов на протяжении первых месяцев жизни также ниже, чем у взрослых. У недоношенных эти процессы более длительные по сравнению с доношенными новорожденными.
Степень связывания антибиотика с белками плазмы, в частности с альбумином, оказывает влияние на его транспорт к тканям, в первую очередь к очагу воспаления. Низкий уровень альбумина, характерный для раннего возраста и недоношенных, резко снижает эффективность антибактериальных препаратов.
Важным фактором биотрансформации антибиотиков является объем экстрацеллюлярной жидкости, который у детей значительно больше, чем у взрослых. У новорожденных объем внеклеточной жидкости составляет 45 %. В течение первых трех месяцев он снижается почти в 1,5 раза. Большинство антибиотиков первоначально распределяются во внеклеточной жидкости, и относительно большой ее объем способен оказать существенное влияние на их фармакодинамику, включая замедление времени достижения пиковой концентрации в крови и более позднее начало терапевтического действия.
Распределение антибиотиков в организме ребенка тесно связано со степенью функциональной зрелости органов выделения, прежде всего почек и печени. Большинство наиболее широко используемых в педиатрии бета-лактамных антибиотиков, в частности цефалоспоринов, экскретируются преимущественно путем клубочковой фильтрации. У новорожденного величина клубочковой фильтрации составляет 1/20–1/30 от таковой взрослого, достигая 3/4 ее к году жизни и выходя на уровень взрослого к 2–3 годам. Становление тубулярной функции почек идет еще более медленными темпами и достигает уровня взрослого к 5–7 годам и позднее. Как результат, удлиняется период полувыведения антибиотиков, что обусловливает необходимость по-стоянного контроля почечных функций ребенка (хотя бы по величине суточного диуреза) с соответствующей корректировкой доз.
Особенности фармакокинетики цефалоспоринов таковы, что в плазме крови, за исключением пероральных препаратов, они прочно связываются с белками. Большинство из них (кроме цефтриаксона, цефотетана, цефоницида и моксалактама) в пределах 1,5–2,5 ч выводятся из плазмы крови в виде активного вещества или в виде метаболитов, обладающих антибактериальной активностью. Ряд препаратов (цефалексин, цефуроксим и др.) способен абсорбироваться в желудочно-кишечном тракте.
Цефалоспорины для парентерального введения могут быть назначены как внутривенно, так и внутримышечно (цефалотин показан только для внутривенного введения). При этом необходимо отметить, что внутримышечное введение большинства цефалоспоринов болезненное, в связи с чем в качестве растворителя рекомендуют использовать лидокаин.
Пероральные цефалоспорины хорошо всасываются в ЖКТ. Биодоступность зависит от конкретного препарата и варьирует от 40 до 95 %. Всасывание цефаклора, цефиксима и цефтибутена может несколько замедляться при наличии пищи. Цефуроксим аксетил во время всасывания гидролизуется с высвобождением активного цефуроксима, причем пища способствует этому процессу.
Цефалоспорины легко проникают в различные ткани и среды организма, включая легкие, органы малого таза, перикард, брюшину, плевру, синовиальные оболочки. С терапевтических позиций большое значение имеет способность ряда цефалоспоринов (цефтриаксон, цефуроксим, цефтазидим, цефотаксим) проникать в цереброспинальную жидкость.
Большинство цефалоспоринов выводятся почками; при этом в моче создаются концентрации этих препаратов, во много раз превышающие минимально ингибирующие для большинства актуальных возбудителей инфекций мочевыводящих путей. Вследствие этого в лечении последних можно с успехом использовать среднетерапевтические дозы цефалоспоринов, но при снижении клубочковой фильтрации необходима соответствующая коррекция вводимой дозы антибиотика. Исключением из этого правила являются цефтриаксон и цефоперазон, экскретируемые преимущественно с желчью. Эти препараты не удаляются при гемодиализе, поэтому при проведении этой процедуры не требуется дополнительного увеличения дозы антибиотика.
При заболеваниях печени, даже в отсутствие асцита и его влияния на распределение антибиотика, существенно нарушается фармакокинетика большинства цефалоспоринов.
В отличие от фторхинолонов и аминогликозидов цефалоспорины не оказывают дозозависимого бактерицидного действия. При приеме препаратов быстро достигается максимальная концентрация их в сыворотке крови с последующим снижением ее ниже минимальной ингибирующей: период полужизни большинства цефалоспоринов составляет 0,5–2 ч, и лишь у цефтриаксона достигает 8 ч. В связи с этим, а также с непостоянным и непродолжительным постантибиотическим эффектом необходимо строго придерживаться рекомендаций относительно кратности введения цефалоспоринов.
Цефалоспорины I поколения
Цефалоридин. Из желудочно-кишечного тракта всасывается плохо, поэтому препарат применяют парентерально. Хорошо проникает в ткани и органы. Наиболее высокая концентрация его в почках и воспаленной мышечной ткани, проникает в амниотическую жидкость и кровь плода. Выводится почками путем клубочковой фильтрации через
24 ч (83 % от введенной дозы). Препарат нефротоксичен и в ряде стран уже не применяется. При нарушении функции почек доза и интервалы между введениями препарата корригируются.
Цефалотин. По фармакокинетике цефалотин аналогичен цефалоридину, однако цефалотин несколько быстрее выводится из организма. В тканях почек и воспаленной мышце его концентрация составляет 100 %, уровень антибио-тика в плевральном, перитонеальном, синовиальном экссудате 50–100 % от уровня его концентрации в крови. Частично выводится печенью, но концентрация его в желчи ниже, чем в крови.
В печени метаболизируется (диацетилируется) с образованием неактивных метаболитов. Почками выводится 65 % препарата в биологически активной форме. Диацетилцефалотин (метаболит цефалотина) сохраняет биологическую активность только в отношении B.subtilis. Нефротоксичность цефалотина менее выражена, чем у цефалоридина.
Цефазолин. Фармакокинетической особенностью цефазолина является более длительное сохранение его терапевтической концентрации в крови, чем у цефалотина (8–12 ч). Наибольшие концентрации препарата при парентеральном введении создаются в тканях печени, почек, легких, в поджелудочной железе, миокарде и других мягких тканях, гное. Концентрация в желчи и желчевыводящих путях превышает таковую в крови. Цефазолин проникает через плацентарный барьер и в молоко матери. Имеются сведения о проникновении в костную ткань. Выводится почками за 24 ч (около 90 %) путем клубочковой фильтрации и канальцевой секреции. Нарушение функции почек приводит к замедлению его выведения.
Цефалексин. Быстро всасывается при приеме внутрь, пероральная абсорбция составляет 90 %. После приема внутрь 250 мг концентрация препарата через 1 и 3 ч равна 9 и 11,6 мг/л, а при применении 500 мг — в 2 раза выше. Через 6 ч выявляется минимальная концентрация, объем распределения
Цефалоспорины II поколения
Цефуроксим. Практически не всасывается из желудочно-кишечного тракта, поэтому применяется парентерально. Максимальная концентрация его при внутривенном введении достигается через 30 мин. Циркулирует в организме на терапевтическом уровне 6 ч и через 12 ч выводится практически полностью. Препарат не подвергается биотрансформации в организме и выводится почти полностью в неизмененном виде почками путем канальцевой и клубочковой фильтрации. За сутки выделяется 85 % введенной дозы. Цефуроксим хорошо проникает в ткани и жидкости, в частности в костную ткань, синовиальную и спинномозговую жидкость.
Цефуроксим аксетил. Является стабильным дериватом цефуроксима для внутреннего применения. Он стабилен к действию бета-лактамаз, вырабатываемых стафилококками, гемофильной палочкой и другими бактериями. Биодоступность при приеме внутрь составляет 50–55 %, а после еды 50–60 %. После приема 500 мг препарата через 2–3 ч наблюдается пик концентрации 7–10 мг/л, а после употребления 250 мг МПК для S.aureus, H.influenzae и B.(Moraxella) catarrhalis сохраняется в течение 2,5–6,6 ч. Период полувыведения (Т1/2) равен 1,8 ч, AUC 40,8, ренальный клиренс 69 мл/мин. Концентрация препарата в мокроте через 1 ч после приема — 3,3 мкг/мл, а спустя 2 ч — 2,8 мкг/мл, что превышает МПК для большинства респираторных патогенов (МПК < 0,125 мкг/мл). До 20 % от концентрации в крови накапливается в миндалинах (1,5 мг/кг).
Цефаклор хорошо адсорбируется после приема внутрь, прием пищи уменьшает всасывание препарата. После приема 250, 500 и 1000 мг препарата концентрация его через 30–60 мин достигает 7, 13, 23 мкг/мл соответственно. В жидкости среднего уха концентрация через 30 мин — 3,8 мкг/мл, а при приеме 250, 500 и 1000 мг содержание в слизистой бронхов 3,8; 4,4; 7,7 мкг/мл, в мокроте концентрация не превышает 0,5 мкг/мл. В первые 2 ч большая часть неизмененного препарата экскретируется с мочой, а всего за 8 ч выводится 60–80 %. При постоянном приеме каждые 8 ч цефаклора в тех же дозах концентрация в моче соответственно 600, 900, 1900 мкг/мл, что определяет его высокую эффективность к инфекции мочевыводящих путей. Т1/2 равен 2,3–2,8 ч, гемодиализ уменьшает его на 25–30 %. Объем распределения составляет
Цефотетан. У здоровых людей 51–81 % препарата экскретируется в неизмененном виде почками в течение 24 ч. При внутривенном введении 1–2 г препарата его концентрация в моче составляет 1700–3500 мкг/мл. Терапевтические концентрации препарата обнаруживаются во многих тканях и жидкостях организма: коже, мышцах, миометрии, эндометрии, яичниках, почках, мочеточниках, мочевом пузыре, миндалинах, желчи, перитонеальной жидкости, амниотической жидкости, шейке матки, пазухах носа.
Цефамандол. При внутримышечном введении наибольшая концентрация цефамандола в крови достигается через 30–120 мин, при внутривенном — через 10 мин. Концентрация препарата начинает снижаться через 4–6 ч. T1/2 составляет 0,5–1 ч. Выводится с мочой, при этом в моче создаются высокие концентрации препарата. Терапевтическая концентрация достигается в плевральной жидкости, желчи, суставах и костях.
Цефокситин. После внутримышечного и внутривенного введения
Лоракарбеф. Препарат хорошо всасывается при приеме внутрь, на его биодоступность не влияют прием пищи, возраст или дисфункция почек.
Цефалоспорины III поколения
Цефотаксим. Цефотаксим хорошо проникает в ткани и жидкости организма. Следует отметить высокую проницаемость его через ГЭБ, что позволяет добиться эффективного лечения менингита. Выводится препарат почками, концентрация антибиотика в моче превышает МПК для чувствительных возбудителей в течение 24 ч, он не нефротоксичен. Цефотаксим ацетилируется в организме и образует два неактивных метаболита — дезацетилцефотаксимлактоны (M2 и M3) и один активный — дезацетилцефотаксим. Максимальная концентрация препарата в сыворотке при внутривенном введении наблюдается через 5 мин, при внутримышечном — через 0,5 ч. При значениях клиренса креатинина менее 5 мл/мин требуется снижение дозы цефотаксима на 50 % с сохранением прежних интервалов введения. После повторных введений препарата тенденция к его кумуляции обнаруживается только у больных с тяжелым нарушением функции почек. Гемодиализ приводит к сильному уменьшению периода полувыведения цефотаксима (на 35 %) и дезацетилцефотаксима (на 53 %) в течение 4–6 ч.
Цефтриаксон. После внутривенного болюсного введения 0,5 и
Цефтазидим. При внутривенном введении T1/2 составляет 1,9 ч. Менее 20 % цефтазидима связывается с белками. Уровень связывания с белками не зависит от концентрации в крови. При внутримышечном введении препарата в дозе 0,5 или
Нарушение функции печени не отражается на фармакокинетике и фармакодинамике препарата у пациентов, которым вводили препарат внутривенно в дозе
Цефоперазон. На 70 % выводится желчными путями, поэтому его доза должна быть меньше при поражении печени. В коррекции дозы при падении клубочковой фильтрации необходимости нет.
Моксалактам. При парентеральном введении моксалактам хорошо проникает в ткани и жидкости организма (в том числе через ГЭБ и в интерстициальную жидкость). Максимальная концентрация его в сыворотке после внутривенного введения достигается через 5 мин. Выделяется препарат с мочой. Коррекция дозы препарата проводится при уровне клубочковой фильтрации менее 50 мл/мин.
Цефтибутен. Фармакокинетика цефтибутена хорошо изучена как у взрослых, так и у детей. Биодоступность как капсул, так и суспензии для детей хорошая. Прием пищи не влияет на всасывание капсул, но на 20 % уменьшает абсорбцию суспензии. Максимальная концентрация в плазме колеблется от 12 до 16 мкг/мл при приеме препарата в дозе 9 мг/кг. Т1/2 в разных возрастных группах колеблется от 2 до 3 ч. 57–70 % препарата в неизмененном виде выделяется с мочой. Цефтибутен хорошо проникает в среднее ухо у детей с острым и хроническим отитом, в другие жидкости: в назальном секрете концентрация составляет 47 % от плазменной, в трахеальном и бронхиальном секретах 50 и 40 % соответственно.
Цефподоксим проксетил является хорошо абсорбируемым при применении внутрь эфиром 3-й генерации цефалоспоринов — цефподоксима. Цефподоксим проксетил относится к предлекарствам (prodrugs), которые в организме (в тонкой кишке) деэстерифицируются, превращаясь в активный метаболит цефподоксим. После однократного и многократного приема препарата от 100 до 400 мг через 1,9–3,1 ч достигается терапевтическая концентрация 1,0–4,5 мг/л. Т1/2 колеблется от 1,9 до 2,8 ч. Абсолютная биодоступность 50 % и может снижаться при приеме пищи, антацидов и блокаторов Н2-гистаминовых рецепторов (циметидина).
В плазме крови на 18–23 % связан с белком, практически не метаболизируется и экскретируется с фекалиями.
Препарат хорошо распределяется в органах и тканях. В легочной ткани в течение 3–6 ч концентрация препарата 0,6–0,9 — 0,5–0,8 мг/кг, что составляет 70–80 % от концентрации в плазме крови, в слизистой бронхов — 0,9 мг/кг (50 %), в альвеолярных клетках — 0,1–0,2 мг/кг (10 %), а в плевральной и воспалительной жидкости накапливается до 70–100 % от концентрации в плазме крови. Концентрация цефподоксима в легочной ткани через 6–8 ч во много раз выше МПК 90 для следующих респираторных возбудителей: M.(B.) catarrhalis — в 2 раза, H.influenzae и S.pneumoniae — в 20 раз, S.pyogenes — примерно в 70 раз. Примерно 90 % цефподоксима после приема экскретируется за 12 ч с мочой. У больных со среднетяжелой и тяжелой почечной недостаточностью Т1/2 удлиняется соответственно до 5,9 и 9,8 ч, а в терминальной стадии его концентрация в крови и Т1/2 увеличиваются в 7 раз.
Цефиксим. Быстро всасывается в пищеварительном тракте. Биодоступность при приеме внутрь составляет около 50 %. Выводится преимущественно с мочой и частично с желчью.
Т1/2 — 3–4 ч. Максимальная концентрация в сыворотке крови достигается через 4 ч (при приеме во время еды на 0,8 ч быстрее). Связывание с белками плазмы крови (в основном с альбуминами) составляет 65 %. Период полувыведения — 3–4 ч. При нарушении функции почек он удлиняется в зависимости от клиренса креатинина: при клиренсе креатинина 20–40 мл/мин Т1/2 составляет 6,4 ч, при 5–10 мл/мин — 11,5 ч. Около 50 % выводится с мочой (преимущественно в неизмененном виде), 10 % — с желчью (табл. 1).

Цефалоспорины IV поколения
Цефепим. При внутривенном введении хорошо распределяется в организме, проникает через ГЭБ. Экскретируется преимущественно в неизмененном виде почками. Т1/2 составляет около 2 ч. В моче, желчи, перитонеальной жидкости, жидкости пузыря, слизистом секрете бронхов, мокроте, простате, аппендиксе и желчном пузыре также достигаются терапевтические концентрации цефепима. В среднем период полувыведения цефепима из организма составляет около 2 часов. У здоровых людей, получавших дозы до
Цефпиром. Из желудочно-кишечного тракта препарат плохо всасывается, но при внутривенном введении быстро проникает в разные органы и ткани; плохо проникает в спинномозговую жидкость. После внутривенного введения сохраняется в крови в терапевтической концентрации в течение 12 ч, что дает основание вводить его 2 раза в сутки. Выделяется в основном почками, в небольших количествах — с желчью.
Цефалоспорины V поколения
Цефтобипрола медокарил. Цефтобипрол на 16 % связывается с белками плазмы, и эта степень связывания не зависит от его концентрации. Биопревращение из пролекарства цефтобипрола медокарила в активное лекарство — цефтобипрол происходит быстро и катализируется эстеразами плазмы. Концентрации пролекарства ничтожно малы, и его удается обнаружить в плазме и моче только во время инфузии. Цефтобипрол подвергается минимальному метаболизму до нециклического метаболита, который микробиологически неактивен. Концентрация этого метаболита ниже концентрации самого цефтобипрола и составляет около 4 % от последней. Цефтобипрол выводится преимущественно в неизмененном виде посредством почечной экскреции, Т1/2 составляет около 3 ч. Главным механизмом почечной экскреции является клубочковая фильтрация, небольшая часть дозы подвергается канальцевой реабсорбции. После введения 1 дозы примерно 89 % ее обнаруживается в моче в виде активного цефтобипрола (83 %), метаболита с открытым кольцом (5 %) и цефтобипрола медокарила (< 1 %).
Фармакокинетика цефтобипрола сходна у здоровых добровольцев и у пациентов с легкой почечной недостаточностью (Cl креатинина 50–80 мл/мин).
Цефтобипрол подвергается минимальному печеночному метаболизму и экскретируется в основном почками в неизмененном виде, и поэтому нет оснований считать, что клиренс цефтобипрола у пациентов с печеночной недостаточностью будет снижен.
Цефалоспорины распределяются во многих тканях, органах (кроме предстательной железы) и секретах. Высокие концентрации отмечаются в легких, почках, печени, мышцах, коже и мягких тканях, костях, синовиальной, перикардиальной, плевральной и перитонеальной жидкостях. В желчи наиболее высокие уровни создают цефтриаксон и цефоперазон. Цефалоспорины, особенно цефуроксим и цефтазидим, хорошо проникают во внутриглазную жидкость, но не создают терапевтических уровней в задней камере глаза.
Способность преодолевать ГЭБ и создавать терапевтические концентрации в СМЖ в наибольшей степени выражена у цефалоспоринов III поколения — цефотаксима, цефтриаксона и цефтазидима, а также цефепима, относящегося к IV поколению. Цефуроксим умеренно проходит через ГЭБ только при воспалении оболочек мозга. При менингитах в случае применения цефотаксима его содержание в ликворе достигает 10–50 % от концентрации в сыворотке крови, цефтриаксона — 20–30 %, цефтазидима — около 20 %, моксалактама — 10 % от уровня в сыворотке.
При почечной недостаточности режимы дозирования большинства цефалоспоринов требуют коррекции. К основным факторам, определяющим необходимость уменьшения дозы антибиотиков при ХПН, относят почечный путь экскреции, высокую степень экскреции вещества в неизменном виде, токсичность препарата.
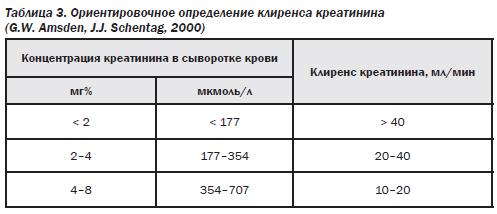
Для коррекции режима дозирования антибиотиков при заболеваниях почек необходимо знать степень нарушения функции почек, которая оценивается по клиренсу креатинина плазмы крови, и степень экскреции препарата почками в неизменном виде (табл. 2).
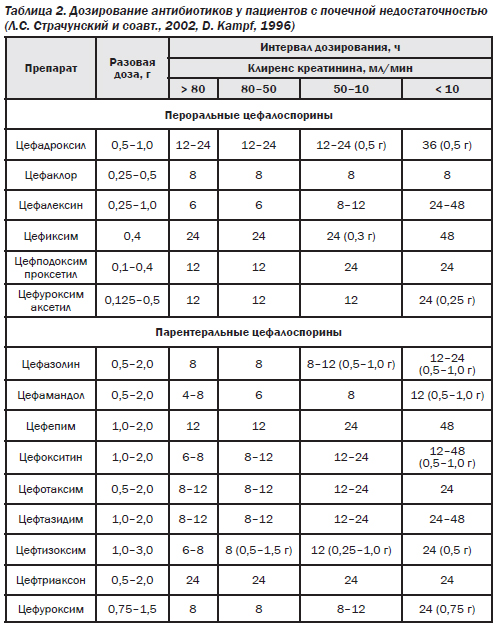
В педиатрии для расчета клиренса креатинина используют формулу Шварца:
КК = [рост (см) / креатинин сыворотки (мкмоль/л) × 0,0113] × к,
дгде к — возрастной коэффициент пересчета:
0,33 — недоношенные новорожденные до 2 лет;
0,45 — доношенные новорожденные до 2 лет;
0,55 — дети 2–14 лет;
0,55 — девочки старше 14 лет;
0,7 — мальчики старше 14 лет.
Возможным является ориентировочное определение клиренса креатинина (табл. 3).
С практической точки зрения важно помнить, что одни из наиболее распространенных в педиатрии пероральных цефалоспоринов — цефуроксима аксетил и цефподоксима проксетил — являются пролекарствами, что значительно повышает их профиль безопасности, в частности минимизирует количество нежелательных гастроинтестинальных явлений.
1. Антибактериальная терапия: Практическое руководство / Под ред. Л.С. Страчунского, Ю.Б. Белоусова, С.Н. Козлова. — М.: РЦ «Фармединфо», 2000.
2. Волосовец А.П., Кривопустов С.П. Цефалоспорины в практике современной педиатрии. — Харьков: Прапор, 2007. — 184 с.
3. Волосовец А.П., Кривопустов С.П., Юлиш Е.И. Антимикробная терапия распространенных заболеваний в дет-ском возрасте. Практическое руководство для врачей. — Киев, 2004.
4. Волосовець О.П., Юліш Є.І. Раціональна антибіотикотерапія респіраторних захворювань у дітей. — Тернопіль: Укрмедкніга, 2003.
5. Майданник В.Г. Проблемы рациональной антибиотикотерапии в педиатрии // Здоров''я України. — 2007. — № 10(167).
6. Посохова К.А., Вікторов О.П. Антибіотики (властивості, застосування, взаємодія): Навч. посібник. — Тернопіль: ТДМУ, 2005.
7. Практическое руководство по антиинфекционной химиотерапии / Под ред. Л.С. Страчунского, Ю.Б. Белоусова, С.Н. Козловa. — М.: Боргес, 2002.
8. Casey J.R. et. al. Metа-analysis of cephalosporin versus penicillin treatment of group A streptococcal tonsillopharyngitis in children // Pediatrics. — 2004. — 113.
9. Nelson Textbook of Pediatrics / Ro-bert M. Kliegman, Richard E. Behrman, Hal B. Jenson, Bonita F. Stanton. — 18th edition. — Saunders, 2007.
10. Red Book: 2006 Report of the committee on Infectious Diseases / Ed. by Larry K. Pickering. — 27th Edition. —