
Журнал «Медико-социальные проблемы семьи» 3-4 (том 17) 2012
Вернуться к номеру
Секвенция Пьера Робена — ранний диагностический признак синдрома Стиклера (клинический случай)

Авторы: Музычина А.А., Малеева И.А. - Донецкий национальный медицинский университет им. М. Горького
Рубрики: Педиатрия/Неонатология, Хирургия
Разделы: Справочник специалиста
Версия для печати
Секвенция Пьера Робена — это симптомокомплекс, включающий триаду признаков: микроретрогнатию, неполную расщелину неба, глоссоптоз. Синдром Стиклера (СС) (наследственная прогрессирующая артроофтальмопатия) характеризуется орофациальными, глазными, ушными, слуховыми и суставными проявлениями. Учитывая позднюю манифестацию СС, выявить патологию у новорожденных затруднительно. Авторы представили семейный случай СС с вариабельными клиническими признаками. Ранняя диагностика СС важна для своевременного выявления его патологических проявлений и определения тактики лечения и ведения пациентов.
Секвенція П’єра Робена — симптомокомплекс, що включає тріаду ознак: мікрогнатію, незрощення піднебіння, глосоптоз. Синдром Стіклера (СС) (спадкова прогресуюча артроофтальмопатія) характеризується орофаціальними, очними, вушними, слуховими й суглобними проявами. Враховуючи пізню маніфестацію СС, виявити патологію в новонароджених важко. Автори описали родинний випадок СС із варіабельними клінічними ознаками. Рання діагностика СС дуже важлива для своєчасного виявлення його патологічних проявів і визначення тактики лікування й ведення пацієнтів.
Pierre Robin sequence — symptom complex that includes a triad of symptoms: microretrognathia, incomplete cleft palate, glossoptosis. Stickler syndrome (SS) (hereditary progressive artrooftalmopatiya) is characterized by orofacial, eye, ear, hearing and articular manifestations. It’s hard to detect pathology in infants taking into account late manifestation of SS. The authors presented the family case of SS with variable clinical features. Early diagnosis of SS is important for timely detection of its pathological manifestations and for determination the tactics of treatment and management of patients.
Орофациальные мальформации, наследственная прогрессирующая артроофтальмопатия.
Орофаціальні мальформації, спадкова прогресуюча артроофтальмопатія.
Оrofacial malformations, hereditary progressive arthroophthalmopathy.
Секвенция (синдром, последовательность, аномалад) Пьера Робена (СПР) — это симптомокомплекс, включающий триаду признаков: микроретрогнатию, неполную расщелину неба или готическое (арковидное, дугообразное) небо, глоссоптоз [1–12].
Французский стоматолог Пьер Робен (1867–1950) в 1923 году на основании 30-летнего наблюдения определил последовательность проявления признаков, которые приводили к обструкции верхних дыхательных путей [2, 3, 5, 7, 9, 12].
Различают СПР изолированную и в составе более 300 синдромов. Изолированная СПР может быть результатом дизрупции/деформации в эмбриогенезе [1–12].
Клиническая картина СПР многообразна, и осложнения различны в разные возрастные периоды жизни ребенка. Ведущим фенотипическим признаком является микрогнатия. Степень выраженности симптомов вариабельна — от незначительной гипоплазии нижней челюсти с арковидным небом, при которой функции жизнеобеспечения не нарушены, дефект носит косметический характер и при своевременной реабилитации не влечет ортодонтических проблем. Тяжелая степень выраженности симптомов ведет к нарушению витальных функций сразу после рождения, в первую очередь к дислокационной асфиксии.
Примерно в 50 % случаев СПР встречаются различные виды расщелин неба: скрытая расщелина неба, расщелина язычка и неполная расщелина. В 10–15 % случаев СПР глоссоптоз сочетается с анкилоглоссией [1–12].
К внечелюстным проявлениям СПР относятся: аномалии ушной раковины и внутреннего уха, глаз, врожденные пороки сердца, патология опорно-двигательного аппарата, мочеполовые мальформации, умственная отсталость [1–12].
В 1965 году американский педиатр Gunnar B. Stickler и соавторы опубликовали результаты длительного наблюдения пациентов с мальформациями орофациальной области, глаз, костно-суставной системы, слуха. Данная патология была определена как прогрессирующая артроофтальмопатия и названа именем автора.
Синдром Стиклера (СС) — аутосомно-доминантная наследственная коллагенопатия (II и IX типы коллагена), имеющая прогрессивное течение и разно-образные проявления в различные возрастные периоды. Популяционная частота 1 : 7500–15 000, соотношение полов (М : Ж) 0,9.
Клинические проявления СС широко варьируют, однако общими симптомами СС являются: орофациальные мальформации (плоское лицо, гипоплазия нижней челюсти, глоссоптоз, арковидное небо и/или различные расщелины неба), глазная патология (миопия, отслойка сетчатки, катаракта, глаукома, аномалия стекловидного тела), дегенеративные заболевания суставов, поражение слуха. Телосложение у пациентов астеническое, грудина воронкообразная, кифоз, сколиоз, вальгусная деформация голеней. Также наблюдаются тугоухость или полная глухота в связи с изменениями во внутреннем ухе.
Манифестация клинических признаков различна и зависит от возраста пациента. Даже в одной семье отмечается значительная вариабельность фенотипов. Развернутая клиническая картина наблюдается после 30-летнего возраста и постоянно прогрессирует. Из общих проявлений синдрома следует отметить увеличение и гиперподвижность суставов у новорожденных и в раннем детстве, в подростковом периоде появляются боль и тугоподвижность в суставах. В юношеском возрасте — прогрессирующий остеоартрит с периодическими обострениями. После 30 лет наблюдается выраженная дегенеративная артропатия тазобедренных, коленных и голеностопных суставов [6, 8–12].
Представляем собственное наблюдение случая СС. Шестимесячный мальчик, рожденный молодыми родителями, без профессиональных и других вредностей, акушерский и семейный анамнезы не отягощены, от первой, нормально протекающей беременности, первых родов, антропометрические показатели соответствовали сроку гестации. Пренатально в сроке 19 недель 5 дней выявлена микрогения, расщелина неба пренатально не диагностирована. Состояние при рождении ребенка тяжелое, нуждался во вспомогательной вентиляции, улучшение состояния отмечалось в положении на животе. У ребенка выявлены микрогнатия, глоссоптоз, расщелина неба, диагностирован синдром Пьера Робена. В периоде новорожденности у ребенка отмечались кислородозависимость, трудности со вскармливанием, патологическая потеря массы тела. После нормализации состояния с рекомендациями по уходу ребенок выписан домой под наблюдение участкового педиатра.
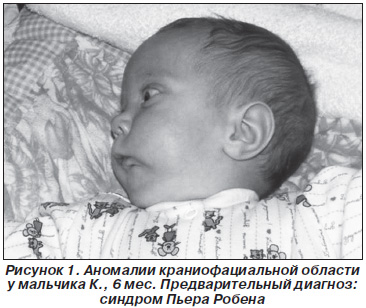
При осмотре в клинике челюстно-лицевой хирургии обращают на себя внимание аномалии краниофациальной области: плоское лицо, микростомия, микроретрогнатия, расщелина твердого и мягкого неба до средней трети. Движения нижней челюсти ограничены. Осмотр ротовой полости затруднен, преддверие уменьшено, неполная расщелина неба до средней трети, дно ротовой полости недоразвито, глоссоптоз (рис. 1). Ребенок обследован: пороки черепно-лицевой области носят изолированный характер. Предварительный диагноз: синдром Пьера Робена.
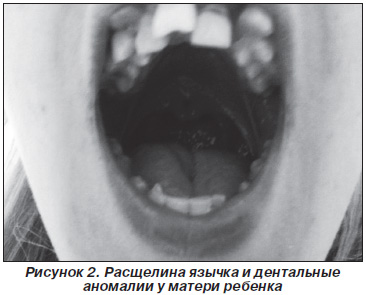
При осмотре у матери ребенка выявлены похожие, но значительно менее выраженные орофациальные проявления: микроретрогнатия, гипоплазия средней трети лица, аномалии зубов, расщелина язычка (рис. 2), при дообследовании диагностирована глазная патология (миопия высокой степени, астигматизм) и суставная патология (незначительные изменения эпифизов локтевых костей).
Из анамнеза: мать также родилась преждевременно. Отмечались трудности со вскармливанием и дефицит массы тела на первом году жизни. С 5-летнего возраста на учете у офтальмолога по поводу близорукости. Кариотип матери 46,ХХ, ребенка 46,ХY.
Дифференциально-диагностический ряд представлен фенотипически подобными синдромами — синдром Марфана, Элерса — Данло, но ключевые признаки данных синдромов отсутствовали. После комплексного обследования матери и ребенка диагностирован синдром Стиклера. При сборе семейного анамнеза не отмечены другие родственники, имеющие признаки артроофтальмопатий. У матери пробанда тип наследования определен как аутосомно-доминантный, мутация de novo.
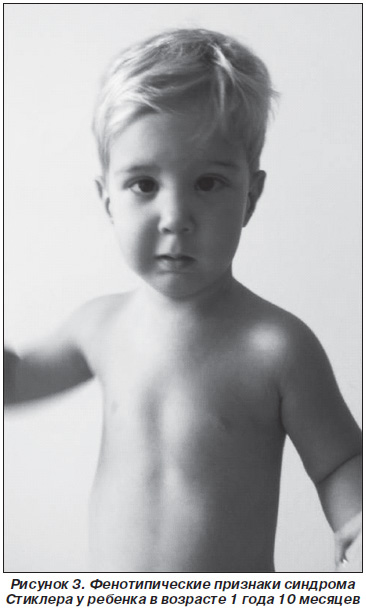
На момент написания статьи мальчику 1 год 10 месяцев. В возрасте 13 месяцев произведена ураностафилопластика. При осмотре у ребенка выявлены: высокое физическое развитие, деформации грудной клетки и конечностей, миопия, астигматизм, страбизм (рис. 3). Однако такой патогномоничный признак, как микрогнатия, регрессирует в результате проводимой комплексной терапии.
Лечение детей с синдромом Пьера Робена поэтапное и длительное, сочетающее консервативные (позиционная терапия, коррекция кормления, терапия сопутствующей патологии) и хирургические методы (трахеостомия, ураностафилопластика и компрессионно-дистракционный остеосинтез). Глоссопексия в настоящее время не применяется. К неотложным мероприятиям относятся устранение обструкции верхних дыхательных путей и коррекция кормления. Ураностафилопластику проводят в возрасте 6–24 месяцев в зависимости от общего состояния пациента. В течение всей жизни, и особенно в раннем возрасте, пациенты должны регулярно наблюдаться у отоларинголога, офтальмолога, ортопеда, логопеда, ортодонта и иммунолога [1, 4, 7–9, 12].
Таким образом, к ранним признакам СС, диагностируемым в периоде новорожденности, относятся лицевые изменения, характерные для СПР, при выявлении которых необходимо дообследование с целью своевременной диагностики и выбора тактики лечения и ведения пациентов. Расщелина неба и более ранние проявления патогномоничных признаков СС можно расценивать как феномен антиципации в данной семье.
1. Хирургическое лечение новорожденных и грудных детей с синдромом Пьера-Робена / Дубин С.А., Комелягин Д.Ю., Злыгарева Н.В. [и др.] // Российский вестник детской хирургии, анестезиологии и реаниматологии. — 2011. — № 2. — С. 33-39.
2. Синдром Пьера Робена у детей / Кириллова Л.Г., Ткачук Л.И., Шевченко А.А. [и др.] // Междунар. неврол. журн. — 2010. — № 3. — С. 18-23.
3. Последовательность Пьера Робена в детской практике / Кириллова Л.Г., Ткачук Л.И., Шевченко А.А. [и др.] // Перинатология и педиатрия. — 2010. — № 2. — С. 32-39.
4. Синдроми вроджених вад, що впливають на зовнішній вигляд обличчя (синдром П’єра-Робена аномалад). МКХ-10 Q 87.0: Протокол надання стоматологічної допомоги // Стоматолог Инфо-Х. — 2010. — № 12. — C. 67.
5. Синдром Пьера Робена в детской практике. Современный взгляд на проблему / О.А. Шевченко // Журнал практичного лікаря. — 2009. — № 1. — С. 29-32.
6. Синдром Стиклера I типа у детей / Семячкина А.Н., Поляков А.В., Новиков П.В. [и др.] // Российский вестник перинатологии и педиатрии. — 2009. — Т. 54. № 3. — С. 45-51.
7. Тактика ведения новорожденных с синдромом Пьера-Робена / Мельникова Е.В., Карачунский М.Г., Тамазян Г.В. [и др.] // Вопросы практической педиатрии. — 2008. — Т. 3. № 5. — С. 36-37.
8. Importance of early diagnosis of Stickler syndrome in newborns / Antunes R.B., Alonso N., Paula R.G. // J. Plast. Reconstr. Aesthet. Surg. — 2012. — Vol. 65. — № 8. — P. 1029-1034.
9. Robin Sequence: From Diagnosis to Development of an Effective Management Plan / Evans K.N., Sie K.C., Hopper R.A. [at al.] // Pediatrics. — 2011. — Vol. 127. — № 5. — P. 936-948.
10. Stickler syndrome, ocular-only variants and a key diagnostic role for the ophthalmologist / Snead M.P., McNinch A.M., Poulson A.V. [at al.] // J. Eye. — 2011. — Vol. 25. — № 11. — P. 1389-1400.
11. Couchouron T. Early-onset progressive osteoarthritis with hereditary progressive ophtalmopathy or Stickler syndrome / T. Couchouron, C. Masson // Joint Bone Spine. — 2011. — Vol. 78. — № 1. — P. 45-49.
12. Lansford M. Focus on the physical assessment of the infant with Stickler syndrome / M. Lansford // Adv. Neonatal Care. — 2008. — Vol. 8. — № 6. — P. 308-314.