
Газета «Новости медицины и фармации» 19 (476) 2013
Вернуться к номеру
Безпека та нормативно-правовий супровід лікарських засобів: від розробки до медичного застосування

Разделы: Медицинские форумы
Версия для печати
23–24 жовтня 2013 року в м. Києві відбулася ІІІ науковопрактична конференція «Безпека та нормативноправовий супровід лікарських засобів: від розробки до медичного застосування», присвячена пам’яті д.м.н., проф. Олексія Павловича Вікторова. Організаторами цього заходу стали Міністерство охорони здоров’я України, Національна академія медичних наук України, ДП «Державний експертний центр МОЗ України» (далі — ДЕЦ). У роботі конференції взяли участь більше ніж 500 учасників: представники ВООЗ, регуляторних органів Великобританії, США, Швеції, Чехії, Білорусі, Російської Федерації, Казахстану, Киргизстану, Вірменії, Молдови, Узбекистану, України, а також керівники управлінь охорони здоров’я, провідні вчені, викладачі медичних і фармацевтичних вузів, представники виробників лікарських засобів (ЛЗ).
Пленарна сесія конференції розпочалася доповіддю Олександра Качура, першого заступника міністра охорони здоров’я України, «Безпека лікарських засобів — складова державної політики в сфері охорони здоров’я». Він розповів про основні напрямки реформування системи охорони здоров’я України, такі як розмежування рівнів надання медичної допомоги, удосконалення надання медичної допомоги матерям і дітям, організація екстреної медичної допомоги, а також розвиток і вдосконалення існуючих механізмів раціональної фармакотерапії.
Регулювання обігу лікарських засобів в Україні, як і в країнах Європи, здійснюється по двох вертикалях, одна з яких контролює якість ліків, друга — здійснює фармаконагляд. Більш детально доповідач зупинився на питаннях, присвячених системі фармаконагляду (ФН) в Україні. Сьогодні це дієва розвинута система, що базується на законодавстві, гармонізованому з європейським, та забезпечує належний нагляд за безпекою ліків Такий підхід має декілька рівнів:
- локальний рівень — це лікувальні заклади, де здійснюється фармакотерапія та відбувається генерація спонтанних повідомлень;
- регіональний рівень представлений регіональними відділами з фармаконагляду, що функціонують у всіх областях України, Автономній Республіці Крим, містах Києві та Севастополі;
- центральний рівень — це департамент післяреєстраційного нагляду ДЕЦ МОЗ України.
Перший заступник міністра відмітив, що система фармаконагляду України неодноразово оцінювалася міжнародними експертами, зокрема, ВООЗ (у 2002 і 2008 рр.). За результатами першої оцінки Україна стала членом Програми ВООЗ з міжнародного моніторингу безпеки лікарських засобів. У 2008 р. вітчизняна система фармаконагляду була визнана кращою серед країн СНД, а в 2012 році за результатами оцінки американських експертів система була визнана кращою серед 40 раніше оцінених країн.
На думку експертів ВООЗ, основним критерієм оцінки системи фармаконагляду є надходження не менше ніж 200 повідомлень про випадки побічних реакцій (ПР) та/або відсутності ефективності (ВЕ) ліків на 1 млн населення. Станом на 2012 р. в Україні рівень інформування про побічні реакції ліків становив 273 повідомлення на 1 млн населення. На сьогодні база даних про побічні реакції лікарських засобів в Україні містить більше ніж 76 тис. спонтанних повідомлень. В основному про побічні реакції повідомляють лікарі. З 2013 року до цього процесу долучилися також пацієнти та фармацевти.
Система фармаконагляду також функціонує у заявників, які виконують, відповідно до законодавства України, вимоги до інформування про несприятливі наслідки застосування лікарських засобів, про результати аналізу співвідношення «користь/ризик».
 За словами Олександра Качура, критеріями оцінки безпеки ліків є частота побічних реакцій, співвідношення кількості непередбачених побічних реакцій до передбачених, серйозних до несерйозних побічних реакцій, виявлення і підтвердження сигналу та співвідношення «користь/ризик». Застосування будьякого лікарського засобу пов’язане з певними ризиками. Покладаючись на спонтанні повідомлення, можна прийняти екстрені регуляторні рішення, такі як: відкликання препарату з ринку, тимчасова заборона або відмова в макетуванні. Але категоричні рішення повинні прийматися тільки в тому випадку, коли співвідношення «користь/ризик» є неприйнятним або ризики стали некерованими, для того щоб не зменшувати користь від застосування ліків для пацієнтів. Доповідач також відмітив, що подальший розвиток системи фармаконагляду в Україні повинен здійснюватися на партнерських взаємовідносинах шляхом:
За словами Олександра Качура, критеріями оцінки безпеки ліків є частота побічних реакцій, співвідношення кількості непередбачених побічних реакцій до передбачених, серйозних до несерйозних побічних реакцій, виявлення і підтвердження сигналу та співвідношення «користь/ризик». Застосування будьякого лікарського засобу пов’язане з певними ризиками. Покладаючись на спонтанні повідомлення, можна прийняти екстрені регуляторні рішення, такі як: відкликання препарату з ринку, тимчасова заборона або відмова в макетуванні. Але категоричні рішення повинні прийматися тільки в тому випадку, коли співвідношення «користь/ризик» є неприйнятним або ризики стали некерованими, для того щоб не зменшувати користь від застосування ліків для пацієнтів. Доповідач також відмітив, що подальший розвиток системи фармаконагляду в Україні повинен здійснюватися на партнерських взаємовідносинах шляхом:
- внесення змін і доповнень до законодавчої бази України в контексті здійснення фармаконагляду;
- посилення виконавчої дисципліни та мотивування лікарів щодо виконання наказів МОЗ України;
- підвищення рівня інформування лікарів з питань безпеки ліків, а також вирішення кадрових і технічних проблем.
Доповідь Олександра Поліщука, співробітника Європейського регіонального бюро ВООЗ, була присвячена взаємозв’язку програм громадської охорони здоров’я і фармаконагляду. Програми громадського здоров’я специфічні для кожної країни та залежать від рівня захворюваності, а також епідеміології поширених захворювань. Дія цих програм спрямована на освіту, зміну навколишнього середовища, корекцію харчування та способу життя населення, а також масове поширення лікарських засобів, проведення вакцинації, лікування і превентивне лікування. Спонсорами програм громадської охорони здоров’я можуть бути ВООЗ, держава та громадські організації, а також приватний сектор.
Програми громадської охорони здоров’я займаються моніторингом захворюваності та поширеності хвороб, смертності, кількості пролікованих хворих та кількості ліків, а як щодо безпеки? Адже застосування будьяких лікарських засобів пов’язане з певними ризиками. Існує також високий ризик неправильного застосування ліків, часто не препарат визначає безпеку, а те, як він застосовується. Тому для виявлення цих ризиків та розробки шляхів із їх мінімізації необхідно впроваджувати фармаконагляд у програми громадського здоров’я. Успіх будьякої програми громадської охорони здоров’я багато в чому залежить від упевненості суспільства в безпеці ліків, а фармаконагляд є основою для раціонального та безпечного використання ліків, тому він повинен стати невід’ємною частиною кожної програми громадської охорони здоров’я.
Досвідом гармонізації вимог до моніторингу безпеки з європейським законодавством поділилась Олена Нагорна, генеральний директор ДЕЦ МОЗ України. Як зазначила доповідач, нормативна база здійснення фармаконагляду в Україні представлена такими документами: Закон України «Про лікарські засоби» 1996 р. зі змінами та доповненнями 1998 р., накази МОЗ України: від 27.12.2006 № 898 «Про затвердження Порядку здійснення нагляду за побічними реакціями ЛЗ, дозволених до медичного застосування в Україні» (гармонізований із директивами ЄС) зі змінами, внесеними наказом від 29.12.2011 № 1005 «Про внесення змін у наказ МОЗ України від 27.12.2006 № 898»; 24.07.2009 № 531 «Про затвердження Порядку здійснення моніторингу безпеки та ефективності ЛЗ у стаціонарах закладів охорони здоров’я»; від 01.09.2009 № 654 «Про затвердження Плану заходів з покращення здійснення післяреєстраційного нагляду за безпекою та ефективністю ЛЗ в стаціонарах закладів охорони здоров’я»; від 31.08.2010 № 736 «Про заходи з впровадження моніторингу безпеки та ефективності ЛЗ у стаціонарах закладів охорони здоров’я»; від 31.10.2012 № 857 «Про своєчасну подачу інформації про випадки побічних реакцій та/або відсутності ефективності ЛЗ працівниками закладів охорони здоров’я»; від 16.08.11 № 595 «Порядок проведення профілактичних щеплень в Україні та контроль якості та обігу медичних імунобіологічних препаратів».
Більш детально доповідач зупинилася на ключових змінах у нормативноправовому полі вітчизняного фармаконагляду. Вона відмітила те, що були розширені поняття «фармаконагляд» та «побічна реакція»; підвищений статус МОЗ України як органу виконавчої влади в системі охорони здоров’я України щодо регулювання питань фармаконагляду; чітко розподілені ролі та обов’язки структур, що беруть участь у процесі обігу лікарських засобів та з питань безпеки; у процесі інформування про побічні реакції, крім лікарів та заявників, залучені медичні сестри, фельдшери, акушери, фармацевти, провізори, споживачі, а також організації, що представляють їх інтереси.
Основні завдання фармаконагляду включають:
- збір інформації про побічні реакції лікарських засобів з усіх доступних джерел та аналіз отриманих даних;
- виявлення та підтвердження можливих нових або змінених ризиків;
- оцінку ризиків; вивчення ризиків через подальші дослідження та управління ризиками шляхом проведення заходів із їх мінімізації;
- оцінку планів управління ризиками, звітів проведених післяреєстраційних (неінтервенційних) досліджень, періодичних звітів із безпеки, звітів з оцінки «користь/ризик»;
- аудит системи фармаконагляду заявників і системи охорони здоров’я;
- забезпечення безпеки застосування лікарських засобів і регулювання обігу ліків шляхом внесення змін в інструкції для медичного застосування препаратів, обмеження або заборона їх застосування.
Обов’язковими елементами системи ФН заявника є наявність у штаті уповноваженої особи, відповідальної за фармаконагляд; наявність структурованої системи фармаконагляду, її оновлення та підтримка; документування всіх процедурних процесів; створення та забезпечення функціонування баз даних із безпеки; забезпечення навчання персоналу компанії для виконання дій, пов’язаних із фармаконаглядом; створення системи управління якістю; а також створення системи управління ризиками у фармаконагляді.
Олена Нагорна також окреслила найближчі задачі, які необхідно здійснити у зв’язку з переміщенням акцентів у фармаконагляді. Ці задачі включають у себе рішення питань кадрових ресурсів, технічного та інформаційного забезпечення, розробку методів та інструментів для оцінки ефективності заходів з управління ризиками, а також оцінки співвідношення «користь/ризик», створення процедур проведення аудиту системи фармаконагляду, розробку ефективних комунікацій і створення вітчизняного керівництва з фармаконагляду.
 Доповідь головного терапевта МОЗ України, чл.кор. НАМН України, д.м.н., професора Василя Нетяженка базувалася на прикладах побічної дії кардіологічних препаратів, зокрема антиангінальних засобів першої лінії.
Доповідь головного терапевта МОЗ України, чл.кор. НАМН України, д.м.н., професора Василя Нетяженка базувалася на прикладах побічної дії кардіологічних препаратів, зокрема антиангінальних засобів першої лінії.
Доповідачем були наведені приклади можливих побічних реакцій залежно від класу препаратів (короткодіючі і тривалодіючі нітрати, блокатори), протипоказання до застосування ЛЗ, міжлікарська взаємодія у контексті механізму дії антиангінальних препаратів.
У доповіді були розглянуті антагоністи кальцію; інгібітори ангіотензинперетворюючого ферменту; діуретики; кордарон щодо можливих побічних реакцій при застосуванні їх, протипоказання до застосування, міжлікарська взаємодія.
Також розглянуті препарати другої лінії, а саме: івабрадин, нікорандил, триметазидин, алопуринол.
Наведений алгоритм антигіпертензивної терапії з використанням основних груп препаратів залежно від показників артеріального тиску (м’яке підвищення, значне підвищення), при цьому зазначалися підстави вибору при їх використанні.
У цьому ж розрізі були розглянуті гіполіпідемічні засоби (статини, фібрати). Надана інформація щодо протипоказань, міжлікарських взаємодій та застережень до застосування таких ЛЗ, як: тромболітики — нефібринспецифічні (стрептокіназа), фібринспецифічні (rtPA); прямі антикоагулянти; нефракціонований гепарин; низькомолекулярні гепарини; непрямі антикоагулянти — варфарин; антитромбоцитарні (аспірин, клопідогрель, тікагрелор, прасугрель і GP2b/3А), та зазначено про те, що об’єднує ці препарати можливе виникнення такої побічної реакції, як кровотечі. Відмічено також, що варіабельність відповіді на клопідогрель залежить від фактора зниження біодоступності клопідогрелю, початкової індивідуальної варіабельності та генетичних особливостей.
Секція 1
була присвячена темі «Інструменти фармаконагляду».
Секцію вели Ульф Бергман, професор фармакоепідеміології кафедри медицини Каролінського інституту м. Стокгольму (Швеція), О.В. Матвєєва, к.м.н., директор департаменту післяреєстраційного нагляду ДП «Державний експертний центр МОЗ України», Т.М. Думенко, к.м.н., директор Департаменту раціональної фармакотерапії та супроводження державної формулярної системи ДП «Державний експертний центр МОЗ України» та О.А. Лебега, старший технічний радник з ФН проект USAID/SIAPS Україна.
Доповідь Ульфа Бергмана була присвячена особливостям застосування ATC/DDDсистеми у фармаконагляді. Доповідач підкреслив, що в 1969 р. в Осло ВООЗ було проведено перший симпозіум «Застосування лікарських засобів». Учасники симпозіуму зазначили гостру необхідність у проведенні досліджень із питань застосування лікарських засобів. Зазначалося, що джерела і форма такої інформації в різних країнах значно відрізнялися.

Для вирішення цієї проблеми була застосована анатомотерапевтична та хімічна (АТХ) класифікація лікарських засобів. АТХкласифікація поділила всі препарати на 5 рівнів залежно від їх дії на певний анатомічний орган або систему, хімічних, фармакологічних та терапевтичних властивостей. Проте для визначення потреби в лікарських засобах важлива наявність не тільки класифікації, але й одиниці виміру. У 1975 р. вчені запропонували нову одиницю — DDD (defined daily dose — встановлена добова доза), під якою розуміють середню підтримуючу дозу лікарського засобу, що застосовується за основними показаннями в дорослих. Було зазначено, що DDD необов’язково повинна відповідати призначеній добовій дозі (prescribed daily dose — PDD). Доза лікарського засобу для одного пацієнта відрізняється від DDD, оскільки залежить від індивідуальних особливостей пацієнта і фармакокінетичних особливостей препарату.
Основним напрямком ATХ/DDDсистеми є дослідження застосування лікарських засобів з метою її вдосконалення. За використання ATХ/DDD методології з’явилася можливість надання статистики застосування як на рівні окремої країни, так і на міжнародному рівні. Однак ця методологія базується тільки на узагальнених даних, не враховує показники захворюваності, тривалості лікування, його ефективність, побічні ефекти й отримані результати.
З доповіддю «Застосування АТХ/DDDметодології для оцінки безпеки основних лікарських засобів» виступила Т.М. Думенко. У доповіді було зазначено, що оцінка безпеки лікарських засобів здійснюється відповідно до об’єктивних критеріїв, серед яких важливе місце займає показник частоти розвитку побічних реакцій.
Розвиток ПР, що часто виникають на окремих територіях та/або у групах населення, може бути пов’язаний з особливостями споживання ЛЗ. У зв’язку з цим Центр у співпраці з Всесвітньою організацією охорони здоров’я зі статистичної методології ЛЗ (м. Осло, Норвегія) рекомендує для отримання об’єктивних показників частоти ПР застосовувати АТХ/DDDметодологію, яка, у свою чергу, базується на використанні Міжнародної АТХкласифікації ЛЗ та дослідженні їх споживання, вимірюваних у встановлених DDD. DDD — розрахункова середня підтримуюча добова доза ЛЗ, що застосовується за основним показанням для дорослого вагою 70 кг.
Використання значень DDD/1000 населення/день чи/або DDD/1000 населення/рік (показників використання ЛЗ) у знаменнику, а кількість побічних реакцій за відповідний період (день, рік) у чисельнику дозволяє дослідити частоту повідомлень про ПР порівняно з обсягами споживання ліків.
Глибина дослідження становила 2008–2012 років. Як вихідні дані використано інформацію з таких джерел: обсяги споживання ЛЗ — дані системи дослідження ринку «Фармстандарт»; кількість спонтанних повідомлень про ПР — національні дані, представлені уповноваженим органом щодо здійснення фармаконагляду в Україні, Департаментом післяреєстраційного нагляду Державного експертного центру МОЗ до Центру співпраці ВООЗ для міжнародного моніторингу ЛЗ (Центр моніторингу м. Упсала, Швеція).
У дослідженні аналізувалась частота розвитку ПР у вигляді всіх порушень із боку різних органів та систем, а також окремо щодо повідомлень про ПР, пов’язаних із порушеннями окремих систем організму.
Обмеженнями до застосування запропонованої методики для оцінки безпеки ЛЗ є такі: узагальнені дані щодо споживання ЛЗ не враховують показників захворюваності та поширеності хвороб; не надають інформації щодо споживання ЛЗ одним пацієнтом; відсутня інформація про результати застосування ЛЗ, неточними є показники кількості випадків у ПР ЛЗ, отриманих методом спонтанних повідомлень, та експозиція пацієнтів, якими використовувався підозрюваний у виникненні ПР ЛЗ.
Також для порівняння окремих ЛЗ необхідно здійснити валідацію за допомогою показника PDD.
Таким чином, отримані результати виявляють загальні тенденції співвідношення кількості повідомлень про ПР та обсягів споживання ЛЗ.
У своїй доповіді Оксана Лебега висвітлила результати аудиту системи фармаконагляду в Україні: сильні та слабкі сторони, напрямки, що потребують розвитку.

У 2011–2012 рр. в Україні відбулася оцінка системи фармаконагляду.
Напрямками та цілями дослідження були:
- Всебічна характеристика та аналіз системи фармаконагляду в Україні, визначення сучасного рівня діяльності.
- Визначення потенційних стратегій зміцнення можливостей системи ФН та їх реалізації.
- Учасниками дослідження були 118 респондентів.
- У доповіді була представлена схема системи ФН, функції та структури спеціалістів (інформаторів та експертів).
- Доповідач представила план дослідження системи ФН та індикатори цього дослідження (ІРАТ). У план включені центральні та регіональні рівні, лікувальні установи та фармацевтична галузь.
- Дослідження охопило всі види ПР ЛЗ, МІБМ та виробів медичного призначення.
Було висвітлено досвід використання IPAT*, а саме:
- Оригінальна або адаптована версія IPAT використовувалась при проведенні досліджень систем ФН у понад 40 країнах світу, включаючи 7 країн Азії.
- З інструментом можна ознайомитись на вебсайті ВООЗ у розділі інструментарію для ФН, користуючись таким посиланням: http://www.pvtoolkit.org/index.php?option=com_content&view=article&id=15&Itemid=19, а також на http://pdf.usaid.gov/pdf_docs/PNADS167.pdf
Дослідження передбачало: аналіз документів та структуровані інтерв’ю з використанням інструменту IPAT (26 основних індикаторів і 17 додаткових).
Досліджувалися такі напрямки:
1. Політики, закони та законодавчі положення.
2. Системи, структури та координація дій зацікавлених сторін.
3. Генерація сигналу та управління даними.
4. Визначення та оцінка ризиків.
5. Управління ризиками та комунікації.
У доповіді надані висновки аудиту:
- Україна досягла суттєвих успіхів у зміцненні національної системи фармаконагляду.
- Міністерство охорони здоров’я та ДЕЦ мають значні досягнення у встановленні базових структур, систем та процесів для покращення безпеки ЛЗ та МІБП.
- Недостатність потенціалу обмежує впровадження діяльності з контролю безпеки ЛЗ та вакцин на обласному рівні та на рівні лікувальних закладів.
- Досягнуто значного прогресу в покращенні спонтанних повідомлень, але діяльність з активного нагляду є дуже обмеженою.
- Існують можливості для активізації фармаконагляду в Державних програмах охорони здоров’я.
- Відсутня юридична база для післяреєстраційного моніторингу безпеки виробів медичного призначення.
У зв’язку із зазначеним ключовими напрямками для розвитку були визначені:
- Розробка національної настанови з ФН та її повне впровадження.
- Розробка та затвердження у МОЗ наказу про проведення аудиту системи ФН у закладах охорони здоров’я (ЗОЗ).
- Включення клінічних провізорів до штатного розкладу ЛУ (у першу чергу протитуберкульозних та центрів СНІДу).
- Розвиток та впровадження навчальних програм із ФН у першу чергу у рамках програм з протидії ТБ.
- Використання можливостей існуючих інформаційних технологій для удосконалення повідомлення про ПР.
- Розробка стратегії збору даних про ПР/ВЕ ЛЗ від пацієнтів.
- Розробка універсального електронного інструменту для здійснення активного ФН.
- Впровадження активного ФН і розробка або з’єднання додаткових методологій та інструментів для оцінки ризиків відповідно (ДПОЗ).
Також доповідач представила Напрямки розвитку ФН при технічній допомозі проекту SIAPS на 2012–2014 рр.
- Розробка національної настанови з ФН, незалежна експертна оцінка нового документа.
- Впровадження посиленого ФН за використання створення інформаційної системи для моніторингу ПР та ВЕ ЛЗ.
- Впровадження активного ФН (пілотного проекту програми протидії ТБ та ВІЛ/СНІДу).
- Розвиток та впровадження тренінгових програм із ФН для програм протидії ТБ та ВІЛ/СНІДу.
- Розвиток та впровадження аудиту ФН у закладах охорони здоров’я та у виробників.
- Розвиток напрямку ФН «Управління ризиками».
Секція 2
мала назву «Нормативноправове забезпечення лікарських засобів»і була спрямована на аналіз нормативноправових документів як в Україні, так і в Польщі.
Головували на секції І.Г. Кудрявцева, д.фарм.н., помічник генерального директора Державного експертного центру МОЗ; Магдалена Владисюк, співробітник Європейського співтовариства технологічної оцінки охороні здоров’я в Центральній і Східній Європі, НТАконсалтинг, Польща; А.В. Степаненко, консультант ДП «Державний експертний центр МОЗ».
Розпочала роботу секції І.Г. Кудрявцева доповіддю «Державна реєстрація лікарських засобів як етап у забезпеченні ефективності, безпечності та якості ліків», у якій зазначила про те, що державна політика у сфері лікарських засобів направлена на забезпечення населення ліками належної якості і в необхідному асортименті, а також забезпечення доступності ліків для соціально незахищених верств населення у випадку погіршення їх здоров’я.
Ірина Георгіївна поінформувала присутніх про те, що державну реєстрацію лікарських засобів здійснює Міністерство охорони здоров’я України на основі результатів експертизи реєстраційних матеріалів, яку проводить Державний експертний центр МОЗ. Основний документ, що регулює відносини, пов’язані зі створенням, реєстрацією, виробництвом та реалізацією лікарських засобів є Закон України «Про лікарські засоби». Даний нормативноправовий документ набув чинності 07.05.1996 р., перетерпів зміни та доповнення. 26.08.2005 р. наказом № 426 Міністерства охорони здоров’я затверджено «Порядок проведення експертизи реєстраційних матеріалів на лікарські засоби, що подаються на державну реєстрацію (перереєстрацію), а також експертизи матеріалів про внесення змін до реєстраційних матеріалів протягом дії реєстраційного посвідчення» (далі — Порядок), який був створений на виконання статті 9 Закону України «Про лікарські засоби» та під час написання Порядку були імплементовані окремі статті Директив ЕС. Остання редакція Порядку була затверджена наказом МОЗ від 04.01.2013 № 3. Зміни до Порядку вводились та будуть вводитись для забезпечення ефективності, безпечності та якості лікарських засобів, тому що цього вимагає сучасне суспільство (синтезуються нові молекули, вдосконалюються методи контролю якості, виявляються нові дії речовин на людський організм та ін.). Доповідач відмітила, що Порядок в останній редакції суттєво відрізняється від редакції восьмирічної давності та відповідає нормативноправовим актам, які прийняті в Європейському Союзі щодо реєстрації лікарських засобів.
Радник міністра охорони здоров’я, консультант ДП «ДЕЦ МОЗ України», д.м.н., професор А.В. Степаненко присвятила свою доповідь ролі медикотехнологічних документів у забезпеченні ефективності та безпеки фармакотерапії. Доповідач розповіла про новини в медикотехнологічних документах, зазначила, що в основу розробок покладені принципи доказової медицини. Уніфіковані клінічні протоколи медичної допомоги і медичні стандарти (УКПМД і МС) розробляються не з медичної спеціальності, а за темою (артеріальна гіпертензія, ішемічний інсульт та ін.). Документи розробляє мультидисциплінарна група (представники всіх спеціальностей, які причетні до різних етапів надання медичної допомоги). У документах вказуються терміни подальшого перегляду. Доповідач відмітила, що публічне обговорення документів (протягом одного місяця знаходяться на сайті МОЗ України). Головне — УКПМД та МС мають індикатори, за якими можна оцінити якість наданої медичної допомоги за певний період, у певному лікувальному закладі, у регіоні, у системі МОЗ України.
У доповіді «Значення безпеки лікарських засобів у звіті Health Technology Assessment» Магдалена Владисюк розкрила значимість безпеки лікарських засобів у звіті Health Technology Assessment (HTA; оцінка медичних технологій). HTA — це комплексна наука, що використовує знання і методи клінічної епідеміології, біостатистики, економіки, права та етики для систематичної оцінки медичних технологій. Головний принцип звіту HTA — чіткий аналіз медичних технологій, прозорість критеріїв для включення або виключення препаратів зі списку лікарських засобів, що підлягають реімбурсації.
Магдаленою Владисюк було також зазначено, що Польща має значний досвід впровадження НТА, що почалося ще в середині 90х років XX ст. У 2009 р. у цій країні створено Агентство з оцінки медичних технологій, яке надає свої поради і думки Міністерству охорони здоров’я, здійснюючи такі завдання:
- Збір, обмін та поширення інформації з оцінки медичних технологій, розроблених у Польщі та інших країнах, їх результати, рекомендації та методи їх підготовки.
- Розробка та верифікація оцінок медичних технологій, і особливо тих технологій, які відпускаються з реімбурсацією.
- Розробка рекомендацій для міністра охорони здоров’я за медичними технологіями.
- Організація навчання та інформування суспільства про свою діяльність.
У доповіді була висвітлена основна мета HTA, яка полягає в тому, щоб дати відповідь на такі питання до здійснення реімбурсації або прийняття цінового рішення щодо конкретної технології:
1. Чи має ця технологія доведену ефективність?
2. У чому сила додаткової корисності в порівнянні з іншими варіантами? (Який з варіантів є найбільш ефективним і в якій мірі він кращий, ніж інші?) Клінічна оцінка, що базується на систематичному огляді.
3. Який із варіантів є найбільш рентабельним і якою мірою він вигідніший, ніж інші? Клінікоекономічний аналіз — це порівняння двох варіантів.
4. Чи виправдане фінансування технології в межах наявних коштів? Якщо технологія отримує привілейоване становище на ринку (наприклад, реімбурсація), з якими змінами це пов’язано?
Роль медикотехнологічних документів у забезпеченні ефективності та безпеки фармакотерапії висвітлила у своїй доповіді А.В. Степаненко.
Секція 3
«Потенціал системи охорони здоров’я в забезпеченні раціональної фармакотерапії»,
яку вели С.М. Дроговоз, Л.С. Савченкова, О.А. Чемет, була спрямована на пошук та вивчення потенціалу системи охорони здоров’я в забезпеченні раціональної фармакотерапії.

Розпочала роботу секції професор кафедри фармакології Національного фармацевтичного університету С.М. Дроговоз доповіддю «Методологічне забезпечення висвітлення питань безпеки лікарських засобів у навчальних програмах», у якій висвітлила методи методичного забезпечення вивчення безпеки ліків в освітніх програмах.
Доповідач акцентувала свою увагу на тому, що основний принцип фармакотерапії був і залишається: користь від ліків повинна перевищувати ризик від їх ускладнень. Тому нагляд за ПР і токсичними проявами ЛЗ та інформація щодо їх запобігання повинні мати першорядне значення в кожній державній програмі, пов’язаній із раціональним використанням ЛЗ. В першу чергу це програми навчання майбутніх лікарів і провізорів, вдосконалення знань лікарів, провізорів, виробників ЛЗ у післядипломному навчанні щодо оцінки співвідношення «користь/ризик» та раціонального застосування ЛЗ, а також з проблем фармаконагляду, надання інформації про нові і, особливо, серйозні ПР ЛЗ.
Світлана Мефодіївна зазначила, що, незважаючи на важливість даної проблеми, універсальних підручників і довідників із ПР, інтоксикації ЛЗ і факторів, що підвищують безпеку ЛЗ, де був би проаналізований, систематизований і уніфікований негативний досвід застосування лікарських засобів, у світі немає. Наявна інформація про ПР ліків та їх передозування в різних джерелах літератури неповна, навіть іноді суперечлива. Джерелами інформації для фахівців про ПР і інтоксикацію, викликану ЛЗ, є підручники, довідники, клінічні керівництва, формуляри, монографії, статті у спеціалізованих періодичних виданнях, бази даних Кокранівського товариства, ВООЗ та ін., інформація від виробників (інструкції для медичного застосування, буклети, брошури, рекламні листки), матеріали науковопрактичних конференцій.
Доповідач відмітила, що за останні 5 років в Україні та Росії видано більше ніж 150 довідників ЛЗ. За 5 років інформація в них про ЛЗ втрачає свою актуальність на 50 %. Найбільш популярними серед фахівців на сьогодні залишаються: «Компендіум», «Лікарські засоби» (М.Д. Машковський), VIDAL (Довідник «Відаль»), «РЛС» (Регістр лікарських засобів Росії).
Підручники, за якими майбутні лікарі та провізори України знайомляться з ПР ліків: за редакцією І.С. Чекмана, 2001 року (рекомендований МОЗ України, обсяг — 600 с.); за редакцією Н.П. Скакуна, 2003 року (рекомендований МОЗ України, обсяг — 740 с.), за редакцією С.М. Дроговоз, 2001 року (рекомендований МОЗ та МОН України, обсяг — 480 с.).
Серед досягнень на сьогодні доповідач зазначила, що дані дисципліни вже є у програмах.
Відповідно до робочої програми навчання на кафедрі фармакології в НФаУ (опорна кафедра) передбачено:
- 72 год (26 год лекцій, 24 год семінарів, 12 год консультацій і 10 год контролю);
- 54 год (10 год лекцій, 20 год лабораторних занять та 24 год самостійної роботи) з токсикології ліків.
Обсяг годин, передбачений у програмах із даних дисциплін становить 1/3 від загальної кількості годин із фармакології (340 год).
Для реалізації цієї робочої програми створені підручники:
- «Побічна дія ліків».
- «Лікарська токсикологія».
- «Хронофармакологія».
- «Фармакологія на допомогу студенту, лікарю і провізору».
- Атлас «Фармакологія — наочно».
На кафедрі фармакології та клінічної фармакології Національного медичного університету ім. О.О. Богомольця розроблений електронний курс «Побічна дія ліків» для студентів 3го курсу, де акцентується увага на ефективно діючій в Україні системі ФН. Доповідач відмітила, що у 2013 році видано методичні рекомендації (МР) «Фармнагляд і його здійснення» (Київ), затверджені МОЗ України від 02.10.2013 р. Розробниками даннях МР є: Державний експертний центр МОЗ України; Національний медичний університет ім. О.О. Богомольця; Донецький національний медичний університет ім. М. Горького і Львівський національний медичний університет ім. Данила Галицького; Бюро ВООЗ. Вісім розділів даних МР (80 с.) присвячені інформації, яку необхідно знати лікарю і провізору для здійснення ФН.
Доповідач зазначила, що Україна однією з перших країн СНД впровадила систему ФН та моніторингу ПР ліків і першою видала свою національну Фармакопею. У 2010 році в Україні видано перший підручник із побічної дії ліків як підручник для студентів вищих навчальних закладів. У 2013 р. видано підручник «Фармакологія на допомогу студенту, лікарю і провізору» за редакцією професорів С.М. Дроговоз, С.Ю. Штриголя, Є.Г. Щокіна. З 900 сторінок даного підручника 230 сторінок (1/4 частина) займає інформація щодо ПР і протипоказань, а також умов, що сприяють і зменшують ПР кожного препарату. Підручник рецензований професорами: І.С. Чекман, В.І. Кресюн, В.І. Мамчур, В.Д. Лук’янчук, Т.В. Звягінцева і рекомендований МОН як підручник для студентів вищих навчальних закладів.
У 2013 році видано атлас «Фармакологія — наочно» під редакцією професора С.М. Дроговоз, у якому логічно представлений взаємозв’язок механізму дії «ЛЗ — ПР — протипоказання» у схемах і таблицях, зручних для розуміння і освоєння особливостей ПР по кожній фармакологічній групі ліків.
До 2013 р. було видано 13 монографій із токсикології ліків.
У 2013 році створений підручник «Лікарська токсикологія» (500 с.) за редакцією професорів С.М. Дроговоз, В.Д. Лук’янчука, Б.С. Шеймана; навчальний посібник «Хронофармакологія — наочно» за редакцією С.М. Дроговоз. Підручник «Хронофармакологія» обсягом 600 с. за редакцією С.М. Дроговоз, С.І. Рапопорта, О.В. Матвєєвої, А.В. Кононенка знаходиться у видавництві. Основним завданням даного видання є надання сучасної хронофармакологічної інформації про підвищення безпеки та ефективності ліків з урахуванням часу їх прийому (діб, сезонів) і можливість зниження вартості проведеної терапії. Це перший підручник з хронофармакології ліків.
На завершення доповідач зазначила, що ПР ліків і лікарська токсикологія не додаток до фармакології, а самостійна галузь пізнання в раціональному застосуванні ліків. Вивчення таких небезпечних властивостей ліків реалізується через селективну інформацію про ПР і токсичності, подану в підручниках «Побічна дія ліків», «Лікарська токсикологія», «Хронофармакологія» і «Фармакологія на допомогу студенту, лікарю і провізору».
Лариса Василівна Савченкова, завідувач кафедри клінічної фармакології та фармакотерапії, д.м.н., професор, від колективу авторів ДЗ «Луганський державний медичний університет» та регіонального відділення з фармаконагляду в Луганській області ДП «Державний експертний центр МОЗ України» представила доповідь «Значення нормативноправових актів і прихильність до їх виконання як основна складова здійснення ФН в Україні».
У своїй доповіді Лариса Василівна дала визначення поняття ФН як виду діяльності, пов’язаного зі збором, визначенням, оцінкою, вивченням та запобіганням виникненню побічних реакцій чи проблем, пов’язаних із застосуванням лікарських засобів.
Система ФН має чітку структуру поетапного збору та оцінки інформації залежно від серйозності проявів побічної реакції та її наслідків. Збір інформації відбувається шляхом отримання спонтанних повідомлень від лікарів закладів охорони здоров’я, моніторингу стаціонарів закладів охорони здоров’я та проведення фармакоепідеміологічних досліджень.
Доповідач зазначила, що протягом 2007–2012 рр. спостерігається стійке зростання отриманих та внесених у базу даних картповідомлень про побічні реакції (далі — ПР) та/або відсутність ефективності ЛЗ. Проте є розбіжність між наданими картамиповідомленнями та внесеними в базу даних. Це обумовлено некоректно внесеними даними щодо підозрюваного лікарського засобу, виробника, заповнення картиповідомлення нерозбірливим почерком тощо.
При аналізі наданих картповідомлень по регіонах України майже всі регіональні відділення подолали рубіж 10 картповідомлень на 100 тис. населення, наданих протягом 2012 р. По Україні цей показник становить 18 картповідомлень на 100 тис. населення. На жаль, невтішні цифри показують, що лише 44 % ЗОЗ протягом 2012 р. та I кварталу 2013 р. брали участь у здійсненні ФН та 50,8 % з них надають 1–3 повідомлення на рік.
За словами доповідача, у регіоні працює 110 ЗОЗ, із них 94 % надають картиповідомлення. Загальна кількість наданих карт від Луганської області у 2012 р. — 728.
Згідно з п. 1.2 наказу МОЗ України від 31.10.2012 р. № 857 «Про своєчасне надання інформації про випадки ПР та/або ВЕ працівниками закладів охорони здоров’я» щопівроку на засіданнях Колегій МОЗ заслухуються питання щодо надання інформації про випадки ПР та/або ВЕ ЛЗ працівниками ЗОЗ у регіонах. У ІІ півріччі 2012 р. у всіх регіонах України було проведено Колегії, де одним із питань було активне залучення ЗОЗ до процесу ФН. Луганська область входить до списку регіонів, де здійснюється посилений контроль з боку керівників структурних підрозділів з питань охорони здоров’я за виконанням наказу МОЗ № 898. За результатами проведеної Колегії спостерігається позитивна динаміка надання картповідомлень з регіонів України.
У доповіді Лариса Василівна розповіла про роботу над виконанням нормативноправових актів МОЗ України в Луганському регіональному відділенні ДП «Державний експертний центр МОЗ України»:
- в установах охорони здоров’я відповідальними за здійснення ФН призначені заступники головних лікарів з лікувальної роботи. При цьому для оптимізації роботи за профілем курації відповідальними призначені терапевти, педіатри, гінекологи, хірурги;
- щотижня на відеоконференціях Департаменту охорони здоров’я Луганської обласної державної адміністрації (далі — ЛОДА) висвітлюються питання моніторингу за ПР ЛЗ, розбираються помилки при заповненні форми 137/о, заслуховуються відповідальні закладів охорони здоров’я про виконання наказу МОЗ України № 898 тощо;
- щоквартально відповідальні здають звіт (форма 69здоров) про кількість поданих повідомлень (наказом МОЗ України № 898 передбачено подання звіту щорічно);
- щоквартально узагальнена інформація щодо поданих повідомлень (у розрізі установ охорони здоров’я області) направляється на адресу керівників закладів охорони здоров’я для можливості вжиття заходів відповідного реагування;
- перевірки в ЗОЗ області виконання наказу МОЗ України № 898, участі лікарів у виявленні та поданні інформації про ПР ЛЗ, складання та виконання локальних протоколів надання медичної допомоги, знання лікарями нормативної бази тощо;
- за результатами перевірки проводяться кущові семінари, на яких за участю фахівців області та представників регіонального відділення обговорюється план роботи з поліпшення моніторингу ПР ЛЗ;
- щомісяця на обласних конференціях Департаменту охорони здоров’я ЛОДА та обласної асоціації терапевтів представниками регіонального відділення висвітлюються питання моніторингу ПР ЛЗ;
- до складу атестаційних комісій з усіх спеціальностей включені представники регіонального відділення;
- у самозвіти лікарів на атестацію за спеціальністю включені розділи індивідуального моніторингу ПР ЛЗ за 3 роки;
- на сайті ЛОДА постійно висвітлюються питання моніторингу ПР ЛЗ;
- виступи представників регіонального відділення на семінарах лікарів приватної медичної практики, зустрічі з представниками громадської організації Товариство Червоного Хреста, ВІЛ/СНІДу, цукрового діабету тощо дозволили підвищити інформованість населення про ПР ЛЗ, можливості подачі інформації в разі її виникнення.
На закінчення свого виступу Лариса Василівна запропонувала шляхи щодо оптимізації здійснення ФН в Україні:
1) посилення виконавчої дисципліни та вмотивованості лікарів щодо виконання наказів МОЗ України з питань здійснення ФН;
2) посилення контролю з боку керівників структурних підрозділів з питань охорони здоров’я обласних, Київської та Севастопольської міських державних адміністрацій за виконанням наказу МОЗ № 898;
3) активізація проведення організаційної та просвітницької роботи регіональних відділень з ФН серед медичних працівників;
4) аудит здійснення ФН у системі охорони здоров’я.
У доповіді О.А. Чемет, головного спеціаліста УЗО Ужгородської ОДА, яка також є представником з ФН Регіонального відділення у Закарпатській області ДП «Державний експертний центр МОЗ України», були висвітлені основи та необхідність проведення аудиту системи ФН у системі охорони здоров’я.
У своїй доповіді О.А. Чемет наголосила, що система ФН забезпечує прийняття відповідних рішень щодо зареєстрованих лікарських засобів на основі отриманої інформації про побічні реакції лікарських засобів (ПР ЛЗ) в умовах їх медичного застосування.
О.А. Чемет нагадала, що методи збору інформації про ПР ЛЗ, які використовуються при здійсненні ФН в Україні на сьогодні є загальновідомими — спонтанні повідомлення, моніторинг стаціонарів, фармакоепідеміологічні дослідження.
Доповідачем детально висвітлена низка факторів, що спричинюють необхідність і водночас підкреслюють важливість інформування про випадки ПР ЛЗ. Вона також звернула увагу на те, про які ПР ЛЗ, у які строки та куди слід повідомляти в разі виникнення побічної реакції.
Причиною недостатньої активності лікарів в інформуванні про ПР ЛЗ, як відмітила доповідач, частіше за все є побоювання, що причиною виникнення ПР/ВЕ ЛЗ стала медична помилка та небажання відповідати за обґрунтованість (або необґрунтованість) проведення фармакотерапії ЛЗ. Побоювання адміністративних стягнень та перевірок щодо надання інформації про ПР та/чи ВЕ ЛЗ, брак часу, формальне ставлення до процесу здійснення нагляду за безпекою ЛЗ.
За даними ДП «Державний експертний центр МОЗ України» у 2012 році лише 44 % ЗОЗ взяли участь у здійсненні ФН, і, наголосила доповідач, лише в 9 регіонах кількість картповідомлень відповідає сучасним критеріям ВООЗ.
Щодо Закарпатського регіону протягом 2012 року до бази даних ДЕЦ надійшли 375 КП (за критеріями ВООЗ — 384). Активність ЗОЗ у здійсненні ФН — 41 %.
Основними кроками до оптимізації здійснення ФН у Закарпатському регіоні, як і в більшості регіонів України, зауважила доповідач, стане залучення всіх медичних працівників всіх ЗОЗ до інформування про випадки ПР та/або ВЕ ЛЗ.
У доповіді наголошено, що з огляду на зазначене вище для оптимізації функціонування системи ФН в Україні необхідно проводити аудит, основними інструментами якого є: законодавче поле; навчання медичного персоналу на регулярній основі питанням безпеки ЛЗ та здійснення ФН; документування усіх процесів у розрізі здійснення ФН, а це стандартні операційні процедури, ведення обліку випадків ПР та/або ВЕ ЛЗ; проведення аналізу щодо активності самого процесу збору інформації та активності медичного персоналу й аналіз власне повідомлень з огляду на якість надання медичної допомоги, відповідність призначень ЛЗ інструкціям для медичного застосування, протоколам надання медичної допомоги та на предмет виявлення медичних помилок.
Не можна відкидати і той факт, наголосила доповідач, що в нашій країні велике значення має адміністративне регулювання. Тому на сьогодні немає більш дієвого механізму оптимізації здійснення ФН, ніж проведення аудиту, критерії контролю якого повинні бути загальновідомими.
Саме тому необхідно МОЗ України створити наказ про затвердження Порядку здійснення ФН у закладах охорони здоров’я, у якому буде чітко прописано порядок здійснення ФН у закладах охорони здоров’я.
Як зазначила доповідач, положення наказу повинні включати:
- визначення термінів;
- загальні вимоги до системи ФН у закладі охорони здоров’я (ЗОЗ);
- загальні вимоги до системи ФН у ЗОЗ;
- загальні вимоги до теоретичної та практичної підготовки працівників з медичною та фармацевтичною освітою з питань ФН, до здійснення ФН у ЗОЗ;
- критерії та підходи оцінки здійснення ФН у ЗОЗ;
- критерії оцінки рівня теоретичної та практичної підготовки працівників закладу охорони здоров’я з медичною і фармацевтичною освітою з питань ФН та підходи до їх оцінок;
- критерії оцінки стану здійснення ФН у закладі охорони здоров’я та їх оціночна шкала.
- порядок проведення аудиту системи ФН у закладі охорони здоров’я та перевірки стану здійснення ФН у закладі охорони здоров’я, теоретичної та практичної підготовки працівників з медичною та фармацевтичною освітою з питань ФН.
За результатами проведення аудиту складатиметься висновок. Під час аудиту встановлюватимуться критичні, суттєві та несуттєві недоліки. У висновку зазначатимуться терміни для їх усунення. А в разі, якщо виявлені недоліки не усуватимуться, МОЗ України вживатиме адміністративні заходи для їх усунення.
Запровадження ефективного медичного аудиту, наголосила доповідач, дасть змогу ефективно використовувати ресурси охорони здоров’я, зокрема: здійснювати об’єктивний аналіз об’ємів наданої медичної допомоги; виявляти та оцінювати недоліки у наданні медичних послуг; комплексно забезпечувати якість її надання; розвивати медичну науку та практику та загалом лобіювати інтереси на користь охорони здоров’я.
Секція 4
була присвячена темі «Розробка ліків, перед і післяреєстраційні дослідження».
Секцію вели О.П. Баула — радник міністра охорони здоров’я України; д.ф.н., професор М.О. Ляпунов — завідувач лабораторією рідких та м’яких лікарських засобів ДП «Державний науковий центр лікарських засобів» (Харків); д.м.н, професор кафедри клінічної фармакології РНДМУ ім. М.І. Пирогова С.К. Зирянов; к.м.н. Л.І. Ковтун — директор Департаменту експертизи матеріалів доклінічних та клінічних випробувань (КВ) Державного експертного центру МОЗ України; д.м.н., професор, заслужений діяч науки і техніки України І.А. Зупанець — завідуючий кафедрою клінічної фармакології з фармацевтичною опікою НФаУ; В.О. Усенко — Dr. Reddy’s Laboratories Ukraine.
У першій частині секції 4 були представлені 2 доповіді.

З доповіддю «Безпека лікарських засобів: фокус на активні фармацевтичні інгредієнти» виступила Ольга Петрівна Баула, радник міністра охорони здоров’я України. У своїй доповіді вона зазначила, що безпека лікарських засобів знаходиться у прямій залежності від фармацевтичних аспектів, одним із яких є якість активного фармацевтичного інгредієнту (АФІ). За визначенням Закону України «Про лікарські засоби», АФІ призначені для використання у виробництві ЛЗ і під час цього використання стають їх активними інгредієнтами, які мають безпосередню дію на організм людини у складі готових лікарських форм. Враховуючи вагомий внесок АФІ в забезпечення якості та безпеки ЛЗ, та з метою запровадження в Україні міжнародних підходів до регулювання обігу АФІ був проведений аналіз тенденцій на ринку цих інгредієнтів, а також норм та положень європейського законодавства, що діє у сфері регулювання АФІ.
Ольга Петрівна підкреслила, що на підставі проведеного аналізу було з’ясовано, що світовий ринок АФІ знаходиться у стані стабільного зростання і склав у 2012 році понад $ 110 млрд США. За прогнозами міжнародних експертів, тенденція до зростання обсягів ринку АФІ буде продовжуватись із щорічним збільшенням у середньому на 5,9 %. Найбільші темпи зростання ринку АФІ будуть спостерігатись у східних країнах, зокрема Індії та Китаї. Вже сьогодні на європейському та американському ринках реалізується понад 80 % ЛЗ, у виробництві яких задіяні АФІ індійського та китайського виробництва. Аналогічна тенденція спостерігається і на фармацевтичному ринку України.
На сучасному етапі розвитку фармацевтичного сектора зазнали суттєвих змін і шляхи постачання АФІ до виробників готових ЛЗ. Безпосередні виробники АФІ здійснюють постачання своєї продукції через складні схеми посередників, брокерів, дистриб’юторів, виробництв з перепакування та перемаркування, що важко контролюються виробниками готових ЛЗ, які несуть відповідальність за їх якість, ефективність та безпеку. На сьогодні існують великі ризики щодо АФІ, а саме:
- постачання АФІ за складними схемами, які часто змінюються, тяжко прослідковуються і є не прогнозованими;
- внесення змін до технологічного процесу та контролю якості АФІ без відома виробника готового ЛЗ;
- неконтрольовані зміни фізикохімічних характеристик АФІ, які можуть мати суттєвий вплив на біодоступність готового ЛЗ;
- неконтрольовані зміни щодо вмісту домішок АФІ, які можуть негативно впливати на безпеку готового ЛЗ;
- застосування підходів до контролю якості АФІ за різними фармакопейними вимогами;
- створення умов для формування «сірого» ринку АФІ;
- створення умов для виробництва та реалізації фальсифікатів АФІ.
Зазначені ризики щодо АФІ призвели до посилення регуляторної політики у сфері забезпечення якості АФІ. В останні роки відбулись суттєві зміни у міжнародних та європейських нормативних актах щодо регулювання виробництва, контролю та постачання АФІ. Відповідно до міжнародних (ICH Q7A, ICH Q11) та європейських (Директива 2001/83/ЄС) вимог виробництво АФІ має здійснюватись тільки згідно з правилами та принципами Належної виробничої практики (GMP). Виробництво готових ЛЗ відповідно до вимог GMP повинно реалізуватись із використанням АФІ, які виготовлені в умовах GMP. Крім того, відповідно до вимог статті 46b Директиви 2001/83/ЄС Європейського Парламенту і Ради ЄС з 02.07.2013 року всі АФІ, які імпортуються до країн Європейського Союзу, повинні мати письмове підтвердження відповідності виробництва АФІ вимогам GMP від уповноваженого органу країни — виробника АФІ. Перед національними уповноваженими органами поставлено відповідальне завдання розробити та запровадити регуляторні функції з інспектування та сертифікації виробництва АФІ. Концепція забезпечення якості готового ЛЗ за міжнародними рекомендаціями має включати елементи управління розробки та виробництва АФІ.
Таким чином, зазначила Ольга Петрівна, для забезпечення безпеки та якості готових ЛЗ в Україні необхідно розробити системний та поетапний підхід до вирішення проблем, що пов’язані з АФІ, а саме:
- створення гармонізованої регуляторної системи щодо АФІ;
- проведення інспектування та сертифікації АФІ;
- впровадження системи надійного постачання АФІ;
- використання АФІ виробниками готових ЛЗ від затверджених постачальників;
- дослідження характеристик АФІ, що впливають на ефективність, безпеку та якість готового лікарського засобу.
Із наступною доповіддю «Сучасні вимоги до фармацевтичної розробки — основа створення якісних, ефективних і безпечних лікарських засобів» виступив завідувач лабораторією рідких та м’яких лікарських засобів ДП «Державний науковий центр лікарських засобів» (м. Харків), доктор фармацевтичних наук, професор Микола Олександрович Ляпунов.
У своїй доповіді Микола Олександрович відзначив, що фармацевтична розробка — це комплексні дослідження, спрямовані на наукове обґрунтування складу лікарського препарату в даній лікарській формі, процесу його виробництва і вибору пакувальних матеріалів, на вивчення фізикохімічних, біологічних і мікробіологічних властивостей, а також сумісності, які слід проводити протягом життєвого циклу продукції з метою створення якісного препарату, його реєстрації та забезпечення якості при серійному виробництві.
Метою фармацевтичної розробки є розробка препарату відповідної якості та процесу його виробництва для подальшого постійного випуску продукції із заданими функціональними характеристиками. Натепер визнано, що якість не може бути повністю перевірено в препаратах, якість має бути закладено при розробці та забезпечено при виробництві.
Професор зазначив, що загальний методологічний підхід до фармацевтичної розробки стандартизований в Керівництві ICH Q8 «Pharmaceutical Development» і в гармонізованому з ним Керівництві СТН МОЗУ 423.0:2011 «Лікарські засоби. Фармацевтична розробка (ICH Q8)», прийнятому МОЗ України.
Згідно з частиною I цих нормативних документів необхідно:
- Дослідити та оцінити компоненти лікарського засобу (лікарські та допоміжні речовини).
- Науково обґрунтувати лікарську форму і склад, включаючи надлишки, дослідити фізикохімічні та біологічні властивості препарату.
- Розробити та оцінити виробничий процес.
- Вибрати систему контейнер/закорковувальний засіб.
- Дослідити мікробіологічні характеристики.
- Визначити, за необхідності, сумісність препарату із розчинниками та/або іншими лікарськими засобами.
Перед початком розробки повинен бути визначений цільовий профіль якості препарату, під яким розуміють очікуваний набір показників, який повинен бути досягнутий для забезпечення необхідної якості препарату з урахуванням його безпеки та ефективності.
Фармацевтична розробка препарату є першою частиною його життєвого циклу й передує організації виробництва. Планування експериментальних досліджень із розробки має здійснюватися з урахуванням оцінки можливих ризиків для якості, що виникають при серійному виробництві.
Микола Олександрович зазначив, що при плануванні фармацевтичної розробки необхідно виробити фармацевтичні та медикобіологічні вимоги до препарату, виділити найбільш значущі фармацевтичні фактори і на підставі результатів експериментальних досліджень оцінити їх вплив на якість препарату, включаючи його функціональні характеристики, конкретні показники якості, ефективність, безпеку та біодоступність. Функціональні характеристики, що визначають якість препарату, специфічні для різних лікарських форм. Це вимагає при розробці кожного препарату специфічних багатофакторних наукових досліджень у взаємозв’язку один з одним, зокрема, скринінгових досліджень в дослідах in vitro та in vivo залежно від значущих фармацевтичних факторів.
Професор навів приклад лікарської форми, для якої функціональні характеристики визначають ефективність дії. Такими лікарськими засобами є препарати для інгаляцій під тиском. Ключовими для ефективності їх дії є такі показники, як аеродинамічний розподіл часток і крапель за розмірами, кількість дрібнодисперсних часток в одній дозі, однорідність доставленої дози. Однак аерозолі різних виробників, для яких декларується одна і та сама доза діючої речовини, можуть відрізнятися за своїми функціональними характеристиками, при цьому вони можуть бути не найкращими у препаратів, які віднесені до референтних.
На відміну від інгаляційних препаратів в аерозольному струмені спреїв з метою безпеки повинна бути виключена або зведена до мінімуму респірабельна фракція частинок. Важливим для безпеки є дозування спреїв. Так, дуже високий ризик побічних ефектів при застосуванні не дозованих спреїв з адреноміметиками, широко представленими на ринку України, у зв’язку з цим раціональними є дозовані спреї, зокрема, на гелевих основах.

Микола Олександрович зауважив, що одним зі шляхів підвищення безпеки лікарської терапії є заміна препаратів для ін’єкцій на препарати системної дії в таких лікарських формах, як назальні спреї та ректальні супозиторії, а також трансдермальні гелі. Їх розробка повинна поєднувати біофармацевтичні, фармакокінетичні та фармакологічні методи досліджень. При цьому важливою функціональною характеристикою, що визначає ефективність і безпеку, може бути осмолярність (осмотична активність), яка повинна відповідати встановленим медикобіологічним вимогам. Це саме стосується препаратів місцевої дії в таких лікарських формах, як мазі, гелі та креми, зокрема, препарати для місцевого лікування ран.
У даний час великою соціальною проблемою є наявність на ринку лікарських засобів, які були розроблені без урахування сучасних стандартів, медикобіологічних вимог та належного наукового обґрунтування. Такі препарати можуть стати причиною неефективності лікування і побічних ефектів, у зв’язку з чим Микола Олександрович навів різні приклади.
Так, наприклад, хондроїтин сульфат натрію через високу молекулярну масу не вивільняється в дослідах in vitro через напівпроникну мембрану, що дозволяє прогнозувати відсутність пенетрації через шкіру. На кафедрі клінічної фармації НФАУ під керівництвом проф. І.А. Зупанця було встановлено, що на моделі системного стероїдного артрозу у щурів хондроїтин сульфат натрію при нашкірному застосуванні у формі мазей, гелів та кремгелів не виявляє хондропротекторної дії. У той же час виражену хондропротекторну активність виявляють кремгелі глюкозаміну гідрохлориду, що відсутні на ринку України.
В Україні і досі триває виробництво стрептоцидової мазі, яка не відповідає сучасним медикобіологічним вимогам і була виключена із номенклатури ще в СРСР через відсутність ефективності та тяжкі побічні ефекти, що виникали при її аплікації на рани.
В Україні виробляють емульсії бензилбензоату з рН близько 10, що створює ризик для стабільності бензилбензоату. Допоміжні речовини не забезпечують фізичну стабільність емульсій, однорідність розподілу бензилбензоату та ефективність антимікробної консервуючої дії.
На думку Миколи Олександровича, не можливо проводити реєстрацію та перереєстрацію неякісних, неефективних та потенційно небезпечних препаратів, що були розроблені за радянських часів, без урахування сучасних стандартів, як препарати «з добре вивченим медичним застосуванням». І ця проблема вимагає свого рішення.
У другій частині секції були представлені 4 доповіді.
Доктор медичних наук, професор кафедри клінічної фармакології РНДМУ ім. М.І. Пирогова Сергій Кенсаринович Зирянов виступив із доповіддю «Післяреєстраційні клінічні дослідження як інструмент оцінки профілю безпеки лікарських засобів».
У своєму виступі Сергій Кенсаринович відмітив, що з моменту розробки препарату до його реалізації можуть знадобитися десятиліття. Доклінічні дослідження тривають від 3 до 5 років (пошук нової діючої речовини, хімічний синтез і початкові дослідження для виявлення активного фармакологічного компонента, вибір лікарської форми та тестування на стабільність).
У І фазі клінічного дослідження (2–3 роки) проводяться випробування нового лікарського засобу (активного компонента) з його попередньою оцінкою. Зазвичай такі випробування проводяться на невеликій групі 20–100 здорових добровольців; відсів: 25 %.
ІІ фаза клінічного дослідження (3–4 роки). Ця фаза вимагає включення більшої кількості добровольців — від 100 до 500 осіб, але із захворюванням або станом, для лікування (діагностики та/або профілактики) якого призначений активний інгредієнт; відсів: 38,8 %.
ІІІ фаза клінічного дослідження (1–2 роки). Випробування проводяться на 1000–3000 пацієнтахдобровольцях, які сплановані для визначення безпеки та ефективності лікарського засобу; відсів: 13,2 %.
ІV фаза клінічного дослідження — постмаркетингові випробування, що проводяться на достатньо великій кількості учасників та протягом тривалого часу для визначення нових режимів прийому препарату, виявлення нових побічних ефектів тощо. Ці дослідження дозволяють отримати більш детальну інформацію про безпеку та ефективність препарату.
Професор зазначив, що розробка 77 % ЛЗ припиняється на стадії клінічних досліджень.
Не тільки в населення, а й у більшості лікарів існує хибна думка з приводу того, що дозволені для застосування в медичній практиці ЛЗ всебічно вивчені та їх безпека з усією повнотою відображена в інструкції про медичне застосування.
Післяреєстраційне клінічне дослідження ЛЗ для медичного застосування проводиться виробником ЛЗ вже після державної реєстрації з метою додаткового збору даних про його безпеку та ефективність, розширення показань до застосування даного ЛЗ, а також виявлення небажаних реакцій пацієнтів на його дію.
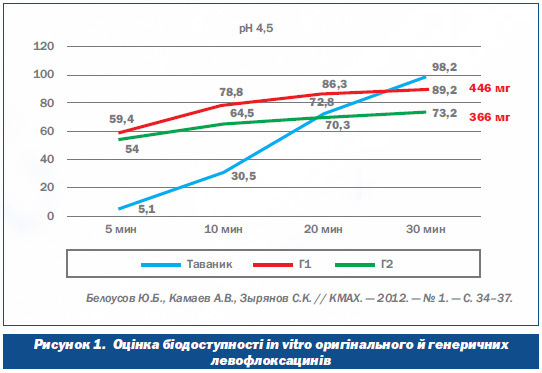
Сергій Кенсаринович навів приклад вивчення еквівалентності показників тесту розчинності оригінального левофлоксацину (таванік виробництва компанії «Санофі», Франція) та двох його генеричних аналогів (Г1 і Г2) з метою прогнозування подібності їх клінічних властивостей. Дослідження кінетики розчинення трьох зазначених лікарських препаратів левофлоксацину проводили при різних значеннях рН середовища. Вибір значень рН середовища ґрунтувався на рекомендаціях Американської фармакопеї. Згідно з цими рекомендаціями, значення рН середовищ, які використовувались у таких дослідженнях, становлять 1,2 і 6,8 для моделювання умов середовища кишкового та шлункового соків і 4,5, як проміжне (оцінка біодоступності in vitro оригінального та генеричних левофлоксацинів). Дослідження показали, що, наприклад, біодоступність при рН середовищі 4,5 оригінального левофлоксацину становить 98,2 % (491 мг), а генериків — 73 % (366 мг) та 89,2 % (446 мг).
Таким чином, зазначив Сергій Кенсаринович, отримані дані дозволяють припустити, що застосування вивчених генеричних аналогів левофлоксацину може супроводжуватися підвищенням ризику клінічних невдач і зростанням резистентності мікроорганізмів до даного препарату.
Ще одним прикладом є дослідження застосування знеболюючих ЛЗ. У дане дослідження були включені 455 амбулаторних хворих із гострим (не менше 7 днів) і хронічним больовим синдромом, з них: 56 % (255 пацієнтів) — чоловіки та 44 % (200 пацієнтів) — жінки. Віковий діапазон становив 18–67 років, середній вік 41,2 ± 8,8 року. У 338 пацієнтів (74,3 %) відмічено больовий синдром у вигляді скелетном’язового болю, який обумовлений дегенеративними або посттравматичними змінами кістковосуглобового і м’язового апарату; у 80 пацієнтів (17,6 %) — у вигляді головного болю напруги.
Виявилося, що в багатьох випадках необґрунтовано призначалися сильнодіючі препарати або препарати offlabel, що призвело до розвитку несприятливих побічних реакцій та впливає на якість життя людини, його фізичне, психологічне та соціальне функціонування.
З доповіддю на тему «Значення клінічних досліджень у вивченні ефективності і безпеки лікарських засобів до етапу реєстрації» виступила директор Департаменту експертизи матеріалів доклінічних та клінічних випробувань Державного експертного центру МОЗ України, к.м.н. Людмила Іванівна Ковтун.
Людмила Іванівна також розповіла про важливість клінічних випробувань у вивченні ефективності та безпеки лікарських засобів до етапу реєстрації, зазначила, які міжнародні нормативні документи регламентують порядок проведення КВ та звітність з безпеки ЛЗ; нормативну базу України, яка регулює проведення КВ в Україні та зупинилась на питаннях подання періодичних звітів із безпеки, у яких щорічно надається періодичний огляд та аналіз інформації з безпеки протягом клінічної розробки лікарського засобу (DSUR).
Доповідач зазначила, що в структурі періодичного звіту з безпеки досліджуваного лікарського засобу (ДЛЗ) має міститися така інформація:
I. Аналіз безпеки:
- Аналіз безпеки і оцінка співвідношення «ризик/користь КВ».
- Нова інформація про безпеку ДЛЗ (враховуються дані про інші лікування та процедури при КВ).
- Внесений у брошуру дослідника або загальну характеристику ДЛЗ (необхідно подавати всю оновлену інформацію, що стосувалася всіх даних із безпеки, не наведених у брошурі дослідника, на початку звітного періоду).
II. Перелік усіх серйозних підозрюваних побічних реакцій, виявлених під час даного КВ, може містити таку інформацію:
- По кожному випадку: код КВ; номер, вік і стать пацієнта в КВ, номер випадку; країна, у якій стався випадок; доза ДЛЗ; дата повідомлення про ПР; дата призначення лікування; опис ПР; його наслідки, коментарі фахівців (зв’язок випадку із ДЛЗ, результати «розсліплення», якщо проводилося).
III. Узагальнені підсумкові таблиці:
- Узагальнені підсумкові таблиці непередбачуваних серйозних побічних реакцій (НСПР) (з їх проявами, симптомами та/або діагнозами).
- Визначається кількість повідомлень: а) для кожної системи організму людини; б) для кожного прояву побічної реакції; в) для кожного періоду та виду лікування.
Результати проведених декількох КВ з одним ДЛЗ наводяться в одному звіті, у якому представлений загальний аналіз безпеки ДЛЗ та аналіз співвідношення «користь/ризик» по кожному КВ.
Людмила Іванівна зазначила, що періодичний звіт із безпеки при КВ служить основою більш якісного проведення КВ шляхом інформування всіх учасників КВ про виявленні ПР/ПЯ; забезпечує своєчасне внесення поправок у брошуру дослідника, протокол КВ та інформовану згоду, своєчасну зупинку КВ при перевищенні ризику над користю для пацієнтів; представляє профіль безпеки ДЛЗ: підсумована інформація про серйозні ризики, що вимагають спостереження з боку виробника та регуляторних органів, також у звіті надається інформація для проведення аналізу з безпеки.
Людмила Іванівна представила розподіл за категоріями звітів із безпеки, отриманих за період з січня 2012 р. по червень 2013 р.: 382 DSUR, 110 переліків ПНСПР, 82 піврічних звіти, 40 інформаційних повідомлень, 20 квартальних звітів, 1 щомісячний звіт.
Загальна кількість позитивних висновків щодо КВ в Україні (1996 р. — вересень 2013 р.) — 3819 КВ: 1996 — 79, 1997 — 90, 1998 — 160, 1999 — 184, 2000 — 182, 2001 — 179, 2002 — 226, 2003 — 249, 2004 — 252, 2005 — 279, 2006 — 322, 2007 — 283, 2008 — 227, 2009 — 184, 2010 — 220, 2011 — 250, 2012 — 265, 9 міс. 2013 — 188.
Міжнародні багатоцентрові КВ (МБКВ) в Україні (2000 р. — вересень 2013 р.): 2000 — 36, 2001 — 36, 2002 — 52, 2003 — 66, 2004 — 85, 2005 — 125, 2006 — 158, 2007 — 185, 2008 — 177, 2009 — 142, 2010 — 177, 2011 — 201, 2012 — 213, 9 міс. 2013 — 140. Затвердження поправок до БМКВ: 2010 — 965, 2011 — 1026, 2012 — 1241, 9 міс. 2013 р. — 911.
На завершення Людмила Іванівна зазначила, що контроль за безпекою ЛЗ проводиться протягом усього «життєвого циклу»:
1. Процес розробки може бути тривалим і не завжди закінчується після реєстрації ЛЗ.
2. Зареєстроване ЛЗ може знову вивчатися в КВ і тоді РSUR і періодичний звіт з безпеки при КВ можуть розроблятися одночасно.
3. Виявлені проблеми безпеки зареєстрованого ЛЗ вимагають повторної оцінки даних КВ, а часом і доклінічних випробувань.
З доповіддю «Дослідження біоеквівалентності як ключовий критерій оцінки безпеки та ефективності генеричних ліків» виступив завідуючий кафедрою клінічної фармакології з фармацевтичною опікою НФаУ, д.м.н., професор, заслужений діяч науки і техніки України Ігор Альбертович Зупанець.
Доповідач відмітив, що постійно зростаючі витрати охорони здоров’я на забезпечення потреб медичного обслуговування населення та ефективне використання коштів — це глобальна міжнародна проблема охорони здоров’я незалежно від політичного та економічного шляху розвитку держави. Саме тому державні органи охорони здоров’я як індустріальних країн і держав, що розвиваються, а останнім часом — і держав з перехідною економікою, заохочують вихід на ринок ліків — аналогів існуючих препаратів, а саме генеричних препаратів. У 2008 р. генеричні препарати становили більш ніж 63 % всіх призначених ліків у США, що привело до зниження фактичних витрат на 13 %. Не стала винятком і Україна, де в останні роки питома вага генериків становить близько 72 %.
Орієнтація на використання генериків, навіть в умовах низької платоспроможності населення, виправдана тільки в тому випадку, якщо вони морально не застаріли і мають доведену еквівалентність оригінальному препарату. Таким чином, основна проблема генерика — доказ подібності оригінальному препарату. Згідно з сучасними вимогами заміна оригінального лікарського засобу на відтворений повинна проводитися тільки в тому випадку, коли генерик відповідає прийнятим міжнародним стандартам, включаючи біоеквівалентність, з метою гарантування якості препаратів. Таким чином, реєстрація генеричного лікарського засобу повинна відбуватися за наявності всіх доказів його терапевтичної взаємозамінності з оригінальним.
Доповідач звернув увагу на те, що неоднозначність у трактуванні результатів і відсутність чітких визначень терапевтичної еквівалентності з боку ВООЗ (Всесвітньої організації охорони здоров’я), FDA (Управління з нагляду за якістю харчових продуктів і лікарських препаратів) та EM (Європейської агенції з лікарських засобів) призводять до невпевненості як лікарів, так і пацієнтів у правильності вибору тих чи інших препаратів генеричного ряду. Тим не менше всі визначення терапевтичної еквівалентності передбачають фармацевтичну еквівалентність або альтернативність і проведення одного з досліджень: вивчення біоеквівалентності на людях (порівняльне фармакокінетичне дослідження); порівняльного дослідження фармакодинаміки на людях; порівняльних клінічних випробувань; тесту розчинності in vitro.
Вибір методу доказу терапевтичної еквівалентності залежить від фізикохімічних властивостей активної субстанції, особливостей фармакокінетики та фармакодинаміки, лікарської форми тощо. На даний момент для таблетованих генериків загальноприйнятим є визнання терапевтичної еквівалентності на основі фармакокінетичної еквівалентності (біоеквівалентності). Тому при виборі генерика можна керуватися тим, що біоеквівалентність лікарських речовин є непрямим підтвердженням їх терапевтичної ефективності. Дослідження біоеквівалентності як методу клінічного випробування дозволяє зробити обґрунтований висновок про ефективність і безпеку порівнюваних препаратів на підставі меншого обсягу первинної інформації і в більш стислі терміни, ніж при проведенні інших видів клінічних випробувань.
Ігор Альбертович Зупанець зазначив, що біоеквівалентність необхідно не тільки вивчати, а й широко висвітлювати отримані дані в наукових статтях, спеціальних довідниках, рекламних матеріалах. У нашій країні необхідна наявність об’єктивної інформації про генеричні препарати, біоеквівалентність яких доведена, як це зроблено в США («Orange Book», Approved Drug Products with Therapeutic Equivalence Evaluation, FDA).
При дотриманні зазначених умов наявність таких даних дозволить повною мірою використовувати взаємозамінні ліки, брати до уваги і економічні переваги, а отже, підвищити якість медичного обслуговування населення.
До уваги учасників конференції була запропонована доповідь «Ефекти плацебо і ноцебо — проблема доказу ефективності і безпеки при проведенні клінічних досліджень», з якою виступив Віталій Олександрович Усенко, Dr. Reddy’s Laboratories Ukraine.
Доповідач зазначив, що за останні 30 років від 10–30 % до 50 % клінічних випробувань не дали доказів ефективності та безпеки нових молекул лікарських засобів (ЛЗ) порівняно з плацебо та існуючими методами лікування. Навіть в онкології частка ЛЗ з ідентичною ефективністю склала майже 25 %. У кардіології і терапії ревматоїдного артриту однакова ефективність щодо існуючих методів лікування спостерігалась у більше ніж 50 % досліджень, а в психіатрії однакова ефективність стосовно існуючих методів лікування виявилася у більше ніж 60 % нових препаратів. З точки зору доказовості 50 % усіх клінічних випробувань у психіатрії не дали бажаного результату. Наприклад, для класу антидепресантів плацебоефект у клінічних випробуваннях становив від 15 до 75 %. Однак подібна проблема характерна не тільки для клінічних випробувань у психіатрії. Так, у середньому від 15 до 30 % клінічних випробувань у всіх терапевтичних галузях не дали позитивних результатів.
В.О. Усенко звернув увагу на те, що, враховуючи актуальність проблем плацебоефектів у клінічних дослідженнях, були проведені аналіз і оцінка аспектів, що впливають на їх якість, вірогідність і можливість подальшого використання у практиці. Проведено огляд та аналіз причин, що призводять до невдач у клінічних випробуваннях. Встановлено типи помилок при плануванні дизайну клінічних випробувань, аналізі даних клінічних випробувань, вплив ефектів плацебо і ноцебо при проведенні клінічних випробувань. Розглянуто сучасні теорії механізму плацебо і ноцебо, факторів, що впливають на посилення ефектів плацебо і ноцебо, включаючи приклади цих ефектів у різних терапевтичних групах та клінічних умовах, розглянуто особливості дизайну клінічних випробувань з мінімізації ефектів плацебо і ноцебо для отримання валідних результатів клінічних випробувань.
Доповідач зазначив, що в подальших дослідженнях будуть запропоновані методичні принципи організації дизайну клінічних випробувань із метою їх оптимізації в напрямку мінімізації ефекту плацебо і ноцебо. Правильна організація дизайну досліджень дозволить збільшити вірогідність даних, отриманих у результаті досліджень, і знизити відсоток невдач при проведенні клінічних випробувань.