
Журнал «Здоровье ребенка» 5 (56) 2014
Вернуться к номеру
Гістіоцитоз із клітин Лангерганса: особливості клініко-лабораторних проявів та перебігу хвороби
Авторы: Дорош О.І. - Західноукраїнський спеціалізований дитячий медичний центр, м. Львів; Цимбалюк-Волошин І.П. - Західноукраїнський спеціалізований дитячий медичний центр, м. Львів; Львівський національний медичний університет імені Данила Галицького; Поліщук Р.С. - Західноукраїнський спеціалізований дитячий медичний центр, м. Львів; Дубей Л.Я. - Львівський національний медичний університет імені Данила Галицького; Воробель О.І., Козлова О.І. - Західноукраїнський спеціалізований дитячий медичний центр, м. Львів; Трояновська О.О. - Західноукраїнський спеціалізований дитячий медичний центр, м. Львів; Львівський національний медичний університет імені Данила Галицького; Степанюк О.І., Скоропад Л.Л. - Західноукраїнський спеціалізований дитячий медичний центр, м. Львів; Кіцера Н.I. - Державна установа «Інститут спадкової патології НАМН України», м. Львів; Середич Л.П., Мих А.М., Грищук Н.Б., Кузьменко А.І. - Західноукраїнський спеціалізований дитячий медичний центр, м. Львів
Рубрики: Педиатрия/Неонатология
Разделы: Клинические исследования
Версия для печати
Мета дослідження. Уточнити особливості клінічних симптомів, перебігу захворювання та ефективності лікування гістіоцитозу з клітин Лангерганса (ГКЛ) у дітей.
Методи дослідження. Клінічний, гематологічний, біохімічний, гістологічний, імуногістохімічний, радіологічний.
Результати дослідження. Проведено аналіз 25 випадків ГКЛ у дітей. Моносистемна форма ГКЛ найчастіше уражає кісткову систему. Мультисистемний ГКЛ характеризується розмаїттям клінічних проявів, значно тяжчим перебігом та високим ризиком смерті. Третину хворих із мультисистемною формою ГКЛ становлять діти першого року життя. Діти з моносистемною формою ГКЛ характеризуються повною клінічною відповіддю на терапію першої лінії. У той же час у дітей із мультисистемною формою повна відповідь на поліхіміотерапію спостерігається лише в 30,0 %. Прогноз перебігу хвороби залежить від ініціального ураження органів ризику (кісткового мозку, печінки, легень, селезінки), порушення їх функції та віку дитини на час встановлення діагнозу. Реактивація процесу в дітей із мультисистемною формою ГКЛ відбувається в перші 12 міс. із часу дебюту хвороби.
Цель исследования. Уточнить особенности клинических симптомов, течения заболевания и эффективности лечения гистиоцитоза из клеток Лангерганса (ГКЛ) у детей.
Методы исследования. Клинический, гематологический, биохимический, гистологический, иммуногистохимический, радиологический.
Результаты исследования. Проведен анализ 25 случаев ГКЛ у детей. Моносистемная форма ГКЛ чаще всего поражает костную систему. Мультисистемный ГКЛ характеризуется многообразием клинических проявлений, более тяжелым течением и высоким риском смерти. Треть больных с мультисистемной формой ГКЛ составляют дети первого года жизни. У детей с моносистемной формой ГКЛ наблюдается полный клинический ответ на терапию первой линии. В то же время у детей с мультисистемной формой ГКЛ полный ответ на полихимиотерапию наблюдается лишь в 30 %. Прогноз течения болезни зависит от инициального поражения органов риска (костного мозга, печени, легких, селезенки), нарушения их функции и возраста ребенка на время установления диагноза. Реактивация процесса у детей с мультисистемной формой ГКЛ происходит в первые 12 мес. со времени дебюта болезни.
The aim of the research. To specify the features of clinical symptoms, course of the disease and the efficacy of treatment for Langerhans cells histiocytosis (LCH) in children.
Methods of the Study. Clinacal, haemotological, biochemical, histological, immunohistochemical, radiological ones.
Results of the Study. An analysis of 25 cases of LCH in children was presented. Monosystem LCH most often affects the skeletal system. Multisystem LCH is characterized by diversity of clinical manifestations, more severe course and high risk of death. One third of patients with multisystem LCH are infants. In children with monosystem LCH we observed complete clinical response to first-line therapy. At the same time, complete response to polychemotherapy is observed only in 30 % of children with multisystem LCH. Prognosis of the disease depends on the initial affection of risk organs (bone marrow, liver, lungs, spleen), their dysfunction and the child’s age at the time of diagnosis. Process reactivation in children with multisystem LCH occurs in the first 12 months from the onset of the disease.
діти, гістіоцитоз із клітин Лангерганса.
дети, гистиоцитоз из клеток Лангерганса.
children, Langerhans cells histiocytosis.
Статья опубликована на с. 40-49
Вступ
Гістіоцитоз із клітин Лангерганса (ГКЛ) — захворювання моноцитарно-макрофагальної системи [37], морфологічним і патофізіологічним субстратом якого є проліферуючі клітини Лангерганса (КЛ). Його різновидами є хвороба Леттера — Зіве, хвороба Хенда — Шюллера — Крісчена, еозинофільна гранульома, хвороба Таратинова, шкірний гістіоцитоз, синдром Хашимото — Притцкера або природжений самовиліковуваний гістіоцитоз, неліпідний ретикулоендотеліоз. Більшість авторів вважають, що всі зазначені одиниці є лише різними клінічними фазами того самого процесу [26, 38]. Захворюваність на ГКЛ становить 2–6 випадків на рік на 1 млн дитячого населення, пік припадає на вікову групу від 0 до 4 років, удвічі частіше хворіють хлопці [3, 36]. В останні роки було доведено, що КЛ при гістіоцитозі є дендритними. Вони разом з лімфоцитами, еозинофілами та нормальними гістіоцитами формують типові для даної патології інфільтрати в різних органах та системах. Етіологія та патогенез хвороби остаточно нез’ясовані. Існують різні теорії походження даної патології: імунологічна, інфекційна тощо [3, 30]. За інфекційною теорією причиною проліферації клітин Лангерганса можуть бути віруси та бактерії, однак відсутність ефекту від противірусної та антибактеріальної терапії робить сумнівною її доцільність. Щоразу частіше привертається увага до генетичного підґрунтя ГКЛ. Описані родинні випадки цього захворювання у двох з чотирьох сибсів [28]. З початку 1980-х років триває створення та постійне вдосконалення програм поліхіміотерапії (ПХТ) ГКЛ. Проте летальність надалі залишається доволі високою. Недостатньо вивченими є також фактори, що впливають на прогноз даної патології [36].
Метою даної роботи стало вивчення особливостей клінічної картини, перебігу та ефективності лікування ГКЛ у дітей. У рамках поставленої мети вирішувались такі завдання: вивчення особливостей клінічної картини і перебігу моносистемної та мультисистемної форм хвороби, оцінка результатів лікування, а також виявлення прогностичних факторів перебігу ГКЛ.
Матеріали і методи дослідження
Проведено аналіз особливостей клініко-лабораторних проявів та перебігу ГКЛ у дітей, які лікувались у відділенні гематології та інтенсивної хіміотерапії КЗ ЛОР «Західноукраїнський спеціалізований дитячий медичний центр» (ЗУСДМЦ) за період з квітня 1992 р. по листопад 2013 р. Діагноз ГКЛ встановлювався на підставі клінічної картини, лабораторних досліджень (загальний аналіз крові, сечі, аналіз сечі за Зимницьким, біохімічний аналіз крові з визначенням загального білка, альбуміну, білірубіну, аланінамінотрансферази, аспартатамінотрансферази, глютамінтранспептидази, лужної фосфатази, загальної лактатдегідрогенази, сечовини, креатиніну, електролітів, глюкози, коагулограми), методів візуалізації (рентгенографія кісток скелета та органів грудної клітки, ультразвукове дослідження (УЗД), комп’ютерна томографія (КТ) та/або магнітно-резонансна томографія (МРТ) голови, органів грудної клітки, черева); пункційної та трепанобіопсії кісткового мозку. Діагноз у всіх аналізованих хворих був підтверджений гістологічним дослідженням біоптату ураженого органа чи тканини (лімфатичний вузол, кісткова тканина, шкіра, ясна, печінка тощо). У частині випадків проведено імуногістохімічне дослідження патологічного субстрату з визначенням CD1a, білка S-100, CD 68, HLA-DR. Терапія проводилась за протоколом СОР та модифікованими програмами DAL-HX-83, –LCH-II, –LCH-III. Статистична обробка результатів виконувалась за допомогою пакету програм Statistica for Windows 8.0 (Statsoft, USA). Функція безподійного виживання EFS (event free survival) розрахована за методом Каплана — Майера. Порівняння виживання між групами — за допомогою Сox’s-F-test. При аналізі безподійного виживання (EFS) подіями вважаються: прогресія, рецидив (реактивація) хвороби, відсутність ремісії повної та часткової після ПХТ 1-ї лінії, смерть хворого.
Результати та їх обговорення
За період з квітня 1992 р. до листопада 2013 р. у відділенні гематології та інтенсивної хіміотерапії ЗУСДМЦ лікувались 25 дітей віком від 2 міс. до 14 років (медіана віку 2 роки 8 міс.), яким діагностовано ГКЛ. Переважали пацієнти віком до чотирьох років (n = 15, 60 %). У семи хворих діагноз встановлено у віці до одного року, що становить 28 % від усієї кількості хворих. Діти віком понад вісім років становили 24 % (n = 6). Співвідношення хлопчиків та дівчаток — 1,78 : 1.
Тривалість діагностики ГКЛ від перших проявів хвороби до встановлення остаточного діагнозу коливалась від одного місяця до трьох років (медіана п’ять місяців). Подовження її тривалості у частини дітей відбувалось унаслідок домінування проявів інтеркурентних захворювань з вираженою інтоксикацією, гіпертермією, втратою маси тіла (n = 6; 24 %), які маскували та ускладнювали діагностику гістіоцитозу.
У багатьох дітей (n = 11; 44 %) ГКЛ дебютував специфічним ураженням шкіри, причому у трьох випадках (12 %) шкірні висипи спостерігалися з народження. Одного хворого (4 %) з одномісячного віку понад рік лікували від корости. У чотирьох (16 %) осіб спостерігався рецидивуючий отит, у двох із них — з періоду новонародженості. Одного чотирнадцятилітнього пацієнта впродовж двох років лікували від гнійного отиту зі зниженням слуху. Один хворий віком 13 років вперше звернувся до лікаря через три роки після появи у нього поліурії та полідипсії. В однієї дитини захворювання дебютувало лімфаденітом, ускладненим вторинною флегмоною шиї.
Тривалим був діагностичний процес у дитини, в анамнезі якої з 3-місячного віку спостерігались рецидивуючі отити, ексудативно-геморагічний дерматит, ентеропатія, рецидивуючий бронхіт та загрозливі для життя інфекційні стани: бактерійно-грибковий сепсис, пневмонія. З віку шість місяців неодноразово перебував на лікуванні в нашій клініці, з діагностичною метою проводились кістково-мозкові пункції, біопсія шкіри, печінки та кісткового мозку. Гістологічні препарати консультовані в Клініці Св. Анни (Відень, Австрія). Даних щодо онкопатології, ГКЛ не виявлено. Дитину обстежували на ВІЛ (СНІД), цитомегаловірусну та Епштейна — Барр інфекції, токсоплазмоз, сифіліс, виключались гістіоцитоз, гемобластоз, мієлодиспластичний синдром, хвороба накопичення, синдром Віскотта — Олдрича (відсутність мутації WASP-гену). У 2-річному віці за клінічними та лабораторними даними — автоімунна гемолітична анемія, автоімунна тромбоцитопенія, гепатомегалія (+ 9 см) спленомегалія (+ 5 см), зниження CD3+-, CD4+- та CD8+-лімфоцитів, нормальні рівні імуноглобулінів, часткова ефективність імнуносупресивної терапії в анамнезі дали підстави встановити діагноз: синдром Еванса. Хворий отримував імуносупресивну терапію: преднізолон, пульс-терапію метилпреднізолоном, внутрішньовенний людський імуноглобулін біовен моно 5%, циклоспорин А впродовж шести місяців. На фоні терапії відзначено нормалізацію показників гемограми, регресію проявів дерматиту, зменшення розмірів печінки (+ 5 см) та селезінки (+ 3,5 см). У віці 3 роки 6 міс. звернувся на плановий огляд, коли виявлено екзофтальм, більше справа, дефект лобної кістки до 1 см, пухлинну інфільтрацію м’яких тканин параорбітальних ділянок, гугнявість голосу. Проведено МРТ голови: у губчастій речовині кісток, що утворюють очну ямку, виявлено білатеральні множинні ізоденсивні до мозкової речовини тканинні маси округлої форми зливного характеру. Процес поширювався на решітчасту та клиноподібну кістки з обтурацією носових ходів, кістки твердого піднебіння, скроневу кістку та зумовлює їх деформацію. Візуалізувалося білатеральне грушоподібне потовщення зорових нервів до 12 мм справа та до 8 мм зліва. Інші відділи мозку — без патології. Офтальмологом зафіксовано застій дисків зорових нервів. У дитини були прояви полідипсії та поліурії, гіпоізостенурія, глюкоза крові в нормі. Виконано біопсію пухлинного утворення в ділянці лобної кістки. Гістологічний висновок: гістіоцитоз із клітин Лангерганса.
Одному хворому, який спостерігався в ортопеда з приводу деструктивного вогнища у правій лопатці, проведено імуногістохімічне дослідження біоптату лопаткової кістки, що виявило CD1a+, S100–, CD45+. Інших системних обстежень не проводилось. Пацієнт отримав опромінення лопаткової кістки у дозі 7 Грей. Через 6 міс. виявлено утворення в ділянці нижньої щелепи зліва. Після комплексного обстеження в гематологічному стаціонарі діагностовано мультисистемний ГКЛ із двобічним ураженням лімфовузлів шийно-підщелепових ділянок, кісткової системи — нижня щелепа, пірамідка правої скроневої кістки, ліва потилична кістка, двобічним отитом.
Для верифікації діагнозу ГКЛ було проведено гістологічне дослідження біоптату шкіри — у 3 (12 %) хворих, кісткового мозку — у 15 (60 %) осіб, лімфатичних вузлів — у 8 (32 %) пацієнтів, кісткової тканини — у 6 (24 %) хворих, ясен — у 2 (8 %) дітей, м’якотканинних пухлинних утворень — у 5 (20 %) хворих. В окремих дітей із мультисистемним ураженням біопсійний матеріал взято з двох різних уражених органів чи тканин.
Імуногістохімічне дослідження проведено в 4 (16 %) хворих. Дендритні клітини експресували CD1a і протеїн S-100, CD68, HLA-DR. При імунофенотипуванні методом флюоресценції на склі відбитків клітин пухлинної маси, лімфатичного вузла або іншого біопсійного матеріалу у 7 (28%) осіб спостерігалась експресія CD1a, CD68, HLA-DR.
Відповідно до ураження патологічним процесом органів і систем хворі були розподілені на три групи. До першої увійшли пацієнти з моносистемною формою ГКЛ (n = 5; 20 %) віком від семи місяців до 13 років 5 міс. (медіана віку 8 років 7 міс.). Другу групу утворили 6 (n = 5) осіб віком від двох місяців до 14 років (медіана віку 2 роки 2 міс.), у яких діагностовано мультисистемний ГКЛ без ураження органів ризику. До третьої групи віднесено 14 (56 %) пацієнтів віком від 2 міс. до 8 років 7 міс. (медіана віку 7 років 8 міс.) із мультисистемним ГКЛ з ураженням органів ризику (ОР+).
Співвідношення хлопчиків та дівчаток у першій групі становило 1 : 4, у другій — 5 : 1, у третій — 2,5 : 1 відповідно.
Відзначено вікові особливості хворих у різних групах ГКЛ. Так, у першій групі з моносистемним процесом переважали діти віком понад чотири роки (80 % хворих), у другій групі з мультисистемним ГКЛ ОР– дітей перших чотирьох років та старше чотирьох років було порівну, у третій групі з мультисистемним ГКЛ ОР+ переважали хворі до чотирьох років, частка яких становила 78,6 %. У віковій категорії дітей першого року життя привертає увагу те, що в шістьох хворих із семи діагностований мультисистемний ГКЛ (85 %), причому у чотирьох — він був ОР+ (57 %). Лише одна дитина першого року життя була віднесена до першої групи моносистемної форми ГКЛ.
Відомо, що моносистемна форма хвороби характеризується ізольованим ураженням кісток скелета у вигляді вогнищ деструкції кісткової тканини літичного характеру або шкіри у вигляді специфічної папульозної, часто з геморагічним компонентом, висипки на тулубі і волосистій частині голови, або ураженням лімфатичних вузлів, що проявляється їх збільшенням [36, 43]. У нашому дослідженні в усіх п’ятьох хворих із моносистемною формою хвороби виявлено лише ураження кісток скелета. Причому у двох (40 %) з них ураження було монофокальним, у трьох (60 %) випадках — поліфокальним. В обох дітей з монофокальним ураженням вогнище деструкції локалізувалось у кістках склепіння черепа розміром 0,7 х 1,5 см в однієї особи та 3,0 х 5,0 см — у другої. У випадках поліфокального моносистемного ураження літичні вогнища були виявлені в кістках черепа, таза, верхніх та нижніх кінцівок, хребта. Ділянки остеодеструкції відзначались різноманіттям анатомічних поєднань: в однієї дитини зафіксовано ураження склепіння гайморової пазухи, кістки таза, хребці Th1, Th4, VII ребро та ліва стегнова кістка, в іншої дитини — стінка орбіти, ліва тім’яна та скронева кістки, ліва ліктьова кістка (середня та нижня третини) та тіло хребця L4 з його патологічним компресійним переломом — у другої дитини. Розміри остеолітичних вогнищ коливались від 0,4 до 1,5 см. Ураження кінцівок супроводжувалось болями та порушенням рухової функції у двох (40 %) хворих. Одинадцятирічна дівчинка з компресійним переломом тіла хребця L4 та кіфозом із вираженим больовим синдромом упродовж 5 міс. потребувала тривалого застосування корегуючого корсета (табл. 1).
/43/43.jpg)
Мультисистемна форма ГКЛ характеризується ураженням двох і більше органів та систем організму, а саме одночасним ураженням кісток, шкіри та лімфатичних вузлів [4, 41, 42]. Окрім вищезгаданих локалізацій процесом можуть уражатись органи ризику — печінка, селезінка, кістковий мозок та легені [8, 41], що у частини хворих супроводжується порушенням їх функції. У нашому спостереженні найчастішим проявом хвороби при мультисистемному ГКЛ було ураження різних кісток черепа, яке виявлено у 23 (92 %) пацієнтів. У кожної п’ятої дитини — остеолітичні вогнища в кістках лицевого скелету. У третини хворих (n = 8; 33,3 %) з ІІ групи та 6 (42,8 %) з ІІІ групи у проекції остеодеструкції були наявні пухлиноподібні м’якотканинні утворення. У 5 (25 %) дітей вогнища деструкції кісткової тканини локалізувались у трубчастих кістках верхніх та нижніх кінцівок, що супроводжувалось болем. В однієї (5 %) особи виявлялось ураження ребер, а у двох (14,2 %) пацієнтів із мультисистемним ураженням зафіксовано літичні зміни у хребцях С5 та L1. По одному випадку ураження реєструвалось у кістках таза (7,1 %) та лопатці (7,1 %) (табл. 1).
Наступними за частотою проявами мультисистемного ГКЛ були: лімфаденопатія — у 14 (56 %) хворих та специфічний отит — у 13 (52 %) осіб. Найчастіше уражались шийні (40 %) та підщелепові лімфатичні вузли (28 %), які утворювали конгломерати. Серед інших локалізацій лімфопроліферації виявлено по одному випадку (4 %) збільшення пахових лімфовузлів та ураження лімфозалоз у середостінні. Ураження шкіри відзначалось у 8 (32 %) дітей переважно у вигляді себорейних та папульозно-петехіальних висипань на шкірі волосистої частини голови, кінцівок, тулуба з утворенням у шкірних складках ерозій та ділянок мокнуття. Інфільтрацію ясен спостерігали у 5 (16 %) пацієнтів, один з яких належав до ІІ групи. У двох дітей із ІІІ групи ГКЛ виявлено некротичний гіперпластичний гінгівіт у поєднанні з остеолітичними вогнищами в нижній щелепі. Екзофтальм при ураженні орбіти діагностовано в 4 (16 %) осіб. У 21,4 % пацієнтів із ІІІ групи мультисистемні ураження поєднувались із проявами нецукрового діабету та у двох хлопців спостерігали низькорослість як прояв специфічної інфільтрації гіпофіза. Слід відзначити, що синдром нецукрового діабету залишається постійною супровідною патологією у хворих, вилікуваних від ГКЛ (табл. 1).
Ураження органів ризику виявлено у 14 (70 %) хворих з мультифокальною формою ГКЛ. Збільшення розмірів печінки від 2 до 10 см спостерігали у всіх дітей, з них у двох (14,2 %) осіб за даними УЗД, КТ чи МРТ діагностовано вогнищеві утворення, в однієї (7,1 %) дитини сонографічно діагностовано інфільтративне ураження судин печінки та нирок. Порушення функції печінки у 5 (35,7 %) хворих проявлялось гіпопротеїнемією з гіпоальбумінемією, у 3 (21,4 %) дітей спостерігалось поєднання гіпопротеїнемії і гіпофібриногенемії, а у 4 (28,5 %) пацієнтів — цитолізу та гіпербілірубінемії. Ураження селезінки у вигляді спленомегалії (від 1,5 до 10,0 см нижче реберної дуги) відзначалось у 8 (57,1%) хворих. Дисфункція гемопоезу у вигляді моно-, бі- або панцитопенії відзначена у 8 (57,1 %) осіб. У половині випадків у мієлограмі виявлено від 10 до 20 % гістіоцитів. У 3 (21,4 %) пацієнтів гістіоцитарна інфільтрація кісткового мозку виявлена при гістологічному дослідженні трепанобіоптату здухвинних кісток. У 3 (21,4 %) дітей ураження кісткового мозку проявлялось трипаростковою цитопенією. У 4 (28,6 %) осіб виявлено специфічну інфільтрацію легеневої тканини, що доведено рентгенологічним та КТ-дослідженнями, у двох із них ураження легень супроводжувалось дихальною недостатністю (табл. 1).
Оскільки досліджувана нами група хворих на ГКЛ формувалась упродовж 21 року, лікування здійснювалось за різними протоколами ПХТ, його результати оцінювали згідно з рекомендаціями міжнародної групи з лікування ГКЛ, а саме: відсутність активної хвороби (відсутні усі клінічні та лабораторні симптоми хвороби), активна хвороба (присутні симптоми ГКЛ) і активна хвороба у стані прогресування (поява нових вогнищ ураження при персистенції чи прогресивному збільшенні первинних) [8, 22] (табл. 1).
Перший хворий із нашого спостереження, якому діагноз мультисистемного ГКЛ ОР– був встановлений у 1992 р., отримав три курси ПХТ за схемою СОР із добрим ефектом. Проте через 24 міс. відзначено реактивацію хвороби. Друга ремісія, досягнута після повторно проведених 3 курсів СОР-терапії, триває упродовж 19,5 року. Лікування за міжнародним протоколом DAL-HX-83 отримали 6 (26 %) хворих, за протоколом LCH-II — 6 (26 %) дітей, за протоколом LCH-III — 9 (39%) хворих (табл. 1).
Лікування в 6 (26,0 %) дітей завершилось летально, з них 4 (17,4 %) дитини отримували терапію за протоколом DAL-HX-83. Причиною смерті було поєднання септичного процесу та прогресування ГКЛ у строках від 0,5 міс. до 6 міс. (медіана 1 міс.) від початку хіміотерапії. Одна (4 %) дівчинка першого року життя з мультисистемною формою ГКЛ з ураженням органів ризику померла від прогресування хвороби, незважаючи на інтенсивну ПХТ із застосуванням двох курсів індукційної терапії за протоколом LCH-III та за протоколом LCH-IІI для прогресуючих мультисистемних уражень 2-хлордезоксіаденозином (2-CdA), а також двох блоків Salvage-терапії СНОР (табл. 2).
/44/44.jpg)
Одна (4 %) дитина у вкрай тяжкому стані внаслідок ураження печінки отримувала лише преднізолон. Однак на 19-ту добу перебування в стаціонарі хворий помер від прогресування ГКЛ. Троє хворих із першої групи отримували лікування за програмою ПХТ, з них одна дитина за протоколом LCH-IІ і дві — за протоколом LCH-IІI. Зокрема, щодо 2 (40 %) дівчаток, які перебували у тривалій ремісії (24 та 51 міс. відповідно), застосовано протокол LCH-IІI. Одному (20 %) хлопчику з поліфокальним ураженням, коли одне з первинних вогнищ локалізувалось в лицевому скелеті, застосовано протокол LCH-IІ для групи В. Зазначимо, що в останнього через 13 місяців з часу первинної діагностики з’явилось нове остеолітичне вогнище у правій стегновій кістці розміром 0,7 х 0,5 х 1,1 см. Дитині проводиться на даний час Salvage-терапія LCH-IІ (блоки А, В). У всіх випадках спостерігалась повна первинна відповідь на лікування. Двоє (40 %) дітей, у яких встановлено моносистемну форму ГКЛ з монофокальним ураженням, не лікувались через відмову батьків. Їх доля невідома (табл. 2).
Добру відповідь на ініціальну терапію у дітей з мультисистемною формою ГКЛ відзначено у 6 (30 %) дітей (з них у одного, який отримав СОР-протоколи, у трьох — протокол LCH-IІ, у двох — протокол LCH-IІІ). В однієї дівчинки з мультисистемним ГКЛ ОР+ через 11 міс. відзначено реактивацію хвороби, що виникла внаслідок відмови матері продовжувати підтримуючу терапію. ІІ лінія терапії була ефективною, пухлина регресувала. Мати дитини через 3 місяці повторно відмовилась від подальшого лікування. Через 25 міс. від припинення терапії відзначено активну прогресуючу хворобу з мультисистемним ураженням із задіянням органів ризику. Застосовано 4 блоки Salvage-терапії LCH-II (А № 3, В № 1) із добрим ефектом. На фоні підтримуючої терапії метотрексатом та пуринетолом через 6 міс. після завершення інтенсивної фази протирецидивного лікування на позитронно-емісійній томографії даних за активну хворобу не виявлено. Ще два підлітки з мультисистемним ГКЛ (один без ураження органів ризику та інший з їх ураженням) продемонстрували добру клінічну відповідь на первинне лікування, однак підтримуюча терапія у них була несистемною, без спостереження гематолога. Через 39 та 13 міс. відповідно у них зафіксовано реактивацію хвороби. На фоні другої лінії Salvage-терапії досягнуто часткову ремісію ГКЛ із персистенцією проявів нецукрового діабету. У зв’язку з досягненням повноліття обидва хворі продовжують лікування у клініці для дорослих. Троє (15 %) дітей із ІІІ групи померли при явищах прогресування ГКЛ унаслідок резистентності захворювання до ПХТ (табл. 2).
Загалом слід відмітити, що реактивація процесу в дітей із мультисистемною формою ГКЛ відбувалася у перші 12 міс. з часу дебюту хвороби.
Кумулятивний відсоток виживання усієї групи хворих за весь період спостереження становив 72 % (рис. 1). Показник безподійного виживання при застосуванні протокольного цитостатичного лікування становив 28 % (рис. 2).
/45/45.jpg)
Тривалість безподійного виживання була статистично вірогідно меншою у дітей до першого року життя порівняно з особами старше 1 року (18 проти 50 %, р = 0,031) (рис. 3). Вірогідно відрізнялись показники виживання у хлопчиків порівняно з дівчатками (12 і 58 % відповідно, р = 0,025) (рис. 4).
/46/46.jpg)
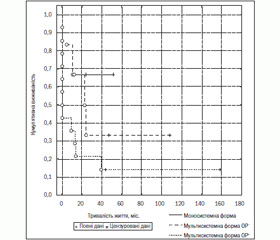
Встановлено, що безподійне виживання було вірогідно гіршим у групі дітей з мультисистемною формою з ОР+ проти виживання у хворих ОР– та з моносистемною формою ГКЛ (14,2 проти 33,3 % та 66,7 % відповідно, р = 0,029) (рис. 5). Слід відзначити, що всі проліковані діти з моносистемною та мультисистемною без ураження органів ризику формами ГКЛ незалежно від виявлення реактивації основного процесу живуть (рис. 6).
/46/46_2.jpg)
З огляду на значну клінічну різноманітність хвороби багато авторів підкреслюють діагностичні труднощі верифікації ГКЛ [20, 21, 26, 30, 39, 43]. За даними нашого дослідження, ГКЛ є доволі рідкісним захворюванням, що зустрічається частіше у хлопчиків віком до 4 років (понад 60 % випадків у нашому спостереженні), що відповідає даним абсолютної більшості публікацій [4, 18, 35, 36]. Поліморфізм клінічних проявів і перебігу хвороби зумовлений тим, що патологічно змінені гістіоцити можуть акумулюватись в одному або багатьох органах, викликаючи порушення їхньої функції. Клініка ГКЛ у нашій вибірці хворих характеризувалася розмаїттям проявів із частим ураженням кісток черепа (92 %), периферичних лімфатичних вузлів (56 %), печінки (40 %), ЦНС (20 %). У понад третини хворих спостерігалась інфільтрація шкіри, селезінки, кісткового мозку та дисфункція ендокринної системи, що підтверджують інші дослідники [1, 12, 43]. За літературними даними [16, 32], ураження зорового аналізатора спостерігається від 1 до 20 % хворих на ГКЛ, що співзвучно з нашими спостереженнями (16 %). У ротовій порожнині ГКЛ може маніфестувати запаленням ясен, болем ясенних сосочків, формуванням нориць, свищів, гранульом у ясенних кишенях. Поширення гранульоми призводить до руйнування кістки та розхитування зубів [21]. У 16 % обстежуваних нами дітей перші ознаки хвороби локалізувались у ротовій порожнині, що проявлялося патологічною рухомістю зубів з наступною їх втратою, кровоточивістю ясен, афтами та гіперплазією ясен. У двох дітей некротичний гіперпластичний гінгівіт супроводжувався сильним болем, що спостерігали інші автори [21, 28]. Рентгенографічно у цих хворих виявлено остеолітичні зміни у щелепних кістках.
Ураження окремих органів та його прогностичний вплив на перебіг ГКЛ різні. За даними міжнародних досліджень загальне виживання пацієнтів високого ризику (мультисистемне ураження із задіянням органів ризику) становить 70 %, а в пацієнтів із відсутністю ранньої відповіді на терапію не перевищує 11–17 % [8, 9, 24]. За результатами нашого дослідження надзвичайно несприятливим прогнозом характеризувались діти з мультисистемною формою ГКЛ із ураженням органів ризику. У пацієнтів із мультисистемним ураженням ОР– найбільшою проблемою є рання реактивація хвороби. Доречно зауважити, що ураження ЦНС, кісток лицевого скелета, передньої або середньої черепної ямки підвищує ризик рецидиву (реактивації) процесу [12]. У нашому дослідженні реактивацію ГКЛ спостерігали в усіх хворих з ураженням лицевого скелета.
Ряд авторів указують, що рання діагностика покращує відповідь на лікування при ГКЛ у дитячому віці [18, 35]. В аналізованих нами хворих досить тривалим був догоспітальний період до встановлення остаточного діагнозу ГКЛ (медіана 5 міс.). Прогноз перебігу ГКЛ залежить також від віку на час встановлення діагнозу, поширеності процесу та ступеня ураження життєво важливих органів. У дітей віком до 1 року перебіг ГКЛ агресивний, зі швидкою дисемінацією процесу, ураженням багатьох органів та їх дисфункцією, з вкрай несприятливим прогнозом [18, 25], що співзвучно з даними нашого дослідження. Отримані результати лікування з безподійним виживанням 28 % можна пояснити тим, що дві третини хворих належали до групи високого ризику, більше ніж половина — за трьома й більше загальноприйнятими критеріями (ураження лицевого скелета, чоловіча стать, гепатоспленомегалія). Окрім того, на результати лікування мали вплив певні незалежні фактори, а саме відмова батьків від протокольного лікування або несистематична терапія. Третина дітей були віком до одного року, вони мали порушення функції життєво важливих органів. Нами діагностовано значно більше випадків інфільтративного ураженням кісткового мозку в дітей (32 %), у той час як окремі автори подають суттєво нижчі цифри — 18 % [39]. Подібно до інших публікацій, вік дітей до одного року на момент встановлення діагнозу асоціювався з поганим прогнозом (рис. 3, р = 0,031). Даним дослідженням підтверджено, що хлопчики мають гірше виживання порівняно з дівчатками, р = 0,025 (рис. 4). Виявлено вірогідно значущу залежність між безподійним виживанням хворих із мультисистемним ураженням із дисфункцією органів порівняно з групою дітей без ураження органів ризику та моносистемною формою (рис. 5, р = 0,029). Описано несприятливий вплив на якість життя перманентних ускладнень ГКЛ, а саме: нецукровий діабет, затримка росту, ортопедичні проблеми, фіброз/цироз печінки, фіброз легенів тощо [13, 27]. Існує припущення, що розвитку таких перманентних ускладнень хвороби сприяють пізній початок лікування й недостатньо інтенсивна терапія [18]. За нашими даними, перманентні ускладнення спостерігались у 8 (32 %), пацієнтів, що проявлялось у 5 (20 %) дітей нецукровим діабетом, а у двох (8 %) випадках — у поєднанні з низькорослістю. Частота нецукрового діабету у хворих на ГКЛ коливається в широких межах: від 5 до 50 % [7]. У 4 (16 %) дітей унаслідок ураження слухового аналізатора розвинулась приглухуватість, в однієї пацієнтки сформувалась псевдомембрана слухового ходу.
Таким чином, незважаючи на успіх сучасної ПХТ, актуальною залишається проблема розробки оптимальної терапії залежно від ступеня ураження та первинної відповіді на лікування. Особливого терапевтичного підходу вимагають діти з мультисистемним ураженням, оскільки при застосуванні стандартної терапії смертність у цій групі є дуже високою.
У ряді публікацій показана ефективність 2-хлордезоксіаденозину (2-CdA) у терапії рефрактерних форм і рецидивів (реактивації) ГКЛ [14, 29, 31, 33, 34]. Нами така терапія застосовувалась лише щодо однієї дитини, проте неуспішно. Описано також застосування циклоспорину А та інтерферону a, антагоністів ФНП-a, талідоміду, 2-дезоксикоформіцину [2, 15, 19, 23]. Сьогодні проведення альтернативної інтенсивнішої ПХТ як терапії першої лінії не пропонується до широкого застосування з огляду на високу гематологічну токсичність і високу частоту життєво загрозливих ускладнень [10, 14, 29, 31, 33, 34].
Трансплантація гемопоетичних стовбурових клітин асоціюється з високим ризиком вісцеральної токсичності і смертності (до 45 %), зумовлених ініціальним ураженням печінки та легень [5, 6, 11, 17]. Однак у зв’язку з невеликою кількістю трансплантованих хворих на ГКЛ оцінити її ефективність на даний час складно [1].
Таким чином, оптимального способу лікування ГКЛ на сьогодні ще не знайдено.
Застосування різноманітних схем хіміотерапії ГКЛ демонструє високу її ефективність у більшості хворих. У той же час залишається група пацієнтів, результати лікування яких залишаються незадовільними. Ця проблема є предметом пошуку на сучасному етапі етіопатогенетичної терапії ГКЛ.
Висновки
ГКЛ є доволі рідкісним захворюванням дитячого віку, що частіше трапляється у хлопчиків перших чотирьох років життя.
Прогностично найсприятливішим є моносистемний ГКЛ, при якому найчастіше уражується кісткова система. Мультисистемна форма характеризується розмаїттям клінічних проявів, значно тяжчим перебігом та вищим ризиком смерті. Несприятливими прогностичними факторами при мультисистемному ГКЛ є ініціальне ураження органів ризику (кісткового мозку, печінки, легень, селезінки) з порушенням їх функції та вік дітей до 1 року на момент встановлення діагнозу.
Реактивація процесу у дітей з мультисистемною формою ГКЛ відбувається у перші 12 міс. з часу дебюту хвороби.
1. Akkari V. Hematopoietic stem cell transplantation in patients with severe Langerhans cell histiocytosis and hematological dysfunction: experience of the French Langerhans Cell Study Group // Bone Marrow Transplant. — 2003. — 31 (12). — 1097-1103.
2. Arceci R., Brenner M., Pritchard J. Controversies and new approaches to treatment of Langerhans cell histiocytosis // Hematol. Oncol. Clin. North. Am. — 1998. — 12 (2). — 339-357.
3. Bhatia S., Nesbit M., Egeler R. et al. Epidemiologic study of Langerhans cell histiocytosis in children // J. Pediatr. — 1997. — 130. — 774-784.
4. Broadbent V., Gadner H. Current therapy for Langerhans cell histiocytosis // Hematol. Oncol. Clin. North. Am. — 1998. — 12. — 327-338.
5. Caselli D., Arico M. The role of BMT in childhood histiocytoses // Bone Marrow Transplant. — 2008. — 41 (2). — 8-13.
6. Conter V., Reciputo A., Arrigo C. et al. Bone marrow transplantation for refractory Langerhans’ cell histiocytosis // Haematologica. — 1996. — 81 (5). — 468-471.
7. Danger D., Broadbent В., Veoman E. et al. The frequency and natural history of diabetes insipidus in children with Langerhans cell histiocytosis // N. Engl. J. Med. — 1989. — 321. — 1157.
8. Gadner H., Grois N., Arico M. et al. A randomized trial of treatment for multisystem Langerhans’ cell histiocytosis // J. Pediatr. — 2001. — 138 (5). — 728-734.
9. Gadner H., Grois N., Potschger U. et al. Improved outcome in multisystem Langerhans cell histiocytosis is associated with therapy intensification // Blood. — 2008. — 111 (5). — 2556-2562.
10. Gadner H., Minkov M., Grois N. et al. Therapy prolongation improves outcome in multisystem Langerhans cell histiocytosis // Blood. — 2013. — 121 (25). — 5006-5014.
11. Greinix H.T., Storb R., Sanders J.E. Marrow transplantation for treatment of multisystem progressive Langerhans cell histiocytosis // Bone Marrow Transplant. — 1992. — 10 (1). — 39-44.
12. Grois N. Central nervous system disease in Langerhans cell histiocytosis // Hematol. Oncol. Clin. North Am. — 1998. — 12 (2). — 287-305.
13. Haupt R., Nanduri V., Calevo M.G. et al. Permanent consequences in Langerhans cell histiocytosis patients: a pilot study from the Histiocyte Society-Late Effects Study Group // Pediatr. Blood Cancer. — 2004. — 42 (5). — 438-444.
14. Imamura T., Sato T., Shiota Y. et al. Outcome of pediatric patients with Langerhans cell histiocytosis treated with 2 chlorodeoxyadenosine: a nationwide survey in Japan // Int. J. Hematol. — 2010. — 91 (4). — 646-651.
15. Jakobson A., Kreuger A., Hagberg H. et al. Sundstrom C. Treatment of Langerhans cell histiocytosis with alpha-interferon // Lancet. — 1987. — 2 (8574). — 1520-1521.
16. Kanavi M., Javadi F, Javadi M. et al. Unifocal Langerhans cell histiocytosis simulating a limbal papilloma // J. Ophtalmic Vis. Res. — 2012. — 7 (3). — 240-243.
17. Kesik V., Citak C., Kismet E., Koseoglu V., Akyuz C. Hematopoietic stem cell transplantation in Langerhans cell histiocytosis: case report and review of the literature // Pediatr. Transplant. — 2009 May. — 13(3). — 371-374.
18. Krstovski N., Janic D., Docmanovic L. et al. Clinical characteristics and survival of children with Langerhans cell histiocytosis // Spr. Arh. Celok. Lek. — 2008. — 136 (9–10). — 514-518.
19. McCowage G., Frush D., Kurtzberg J. Successful treatment of two children with Langerhans’ cell histiocytosis with 2’-deoxycoformycin // J. Pediatr. Hematol. Oncol. — 1996. — 18 (2). — 154-158.
20. Melanud A., Efrat M., Sova Y. Epibular Module as a Symptom of Langerhans cell histiocytosis // Arch. Ophtalmol. — 2002. — 120 (10). — 1400-1401.
21. Minguez I., Minguez J., Bonet J. et al. Oral manifestations of chronic disseminated histiocytosis. A report of 10 cases // Med. Oral. — 2004. — 9. — 149-154.
22. Minkov M. Treatment of multisys tem Langerhans cell histiocytosis. Results of the DALHX 83 and DALHX 90 studies. DAL HX Study Group // Klin. Padiatr. — 2000. — 212 (4). — 139-144.
23. Minkov M., Grois N., Broadbent V. et al. Cyclosporine A therapy for multisystem langerhans cell histiocytosis // Med. Pediatr. Oncol. — 1999. — 33 (5). — 482-485.
24. Minkov M., Grois N., Heitger A. et al. Response to initial treatment of multisystem Langerhans cell histiocytosis: an important prognostic indicator // Med. Pediatr. Oncol. — 2002. — 39 (6). — 581-585.
25. Mosterd K., van Marion A., van Steensel M. Neonatal Langerhans cell histiocytosis: a rare and potentially life-threatening disease // Int. J. Dermatol. — 2008. — 47 (1). — 10-12.
26. Perek D. Histiocytosis X u dzieci w świetle własnych obserwacji // Ped. Pol. — 1982. — 7–8. — 555-564.
27. Pollono D., Rey G., Latella A. et al. Reactivation and risk of sequelae in Langerhans cell histiocytosis // Pediatr. Blood. Cancer. — 2007. — 48 (7). — 696-699.
28. Rapp G., Motta A Periodontal Disease Associated with Langerhans’ Cell. Histiocytosis: Case Report // Braz. Dent. J. — 2000. — 11 (1). — 59-66.
29. Saven A., Foon K., Piro L. 2-Chlorodeoxyadenosine-induced complete remissions in Langerhans-cell histiocytosis // Ann. Intern. Med. — 1994. — 121 (6). — 430-437.
30. Skierski T., Welfel L., Grzybowski W. Problematyka histiocytosis X w laryngologii // Otolaryngol. Pol. — 1994. — 48 (18). — 347-351.
31. Stine K., Saylors R., Williams L. et al. 2-Chlorodeoxyadenosine (2-CDA) for the treatment of refractory or recurrent Langerhans cell histiocytosis (LCH) in pediatric patients // Med. Pediatr. Oncol. — 1997. — 29 (4). — 288-292.
32. Vosoghi H., Rodriguez-Galindo C., Wilson M. Orbital involutment in Langerhans cell histiocytosis // Ophthal. Plast. Reconstr. Surg. — 2009. — 25. — 430-433.
33. Weitzman S., Braier J., Donadieu J. et al. 2’-Chlorodeoxyadenosine (2-CdA) as salvage therapy for Langerhans cell histiocytosis (LCH). Results of the LCH-S-98 protocol of the Histiocyte Society // Pediatr. Blood. Cancer. — 2009. — 53 (7). — 1271-610.
34. Weitzman S., Wayne A., Arceci R. et al. Nucleoside analogues in the therapy of Langerhans cell histiocytosis: a survey of members of the histiocyte society and review of the literature // Med. Pediatr. Oncol. — 1999. — 33 (5). — 476-481.
35. Валиев Т., Махонова Л., Ковригина А. и др. Случай врожденного лангергансоклеточного гистиоцитоза у ребенка раннего возраста // Онкогематология. — 2011. — 2. — 19-22.
36. Коколина В., Румянцев А. Практическое руководство по детским болезням. — М.: Медпрактика-М, 2004. — Т. 4. — 578, 581, 586.
37. Луговская С., Лукина Е., Цветаева Н.В. Морфофункциональная характеристика мононуклеарных фагоцитов лейкоконцентрата венозной крови больных гистиоцитозами // Тер. архив. — 1994. — 4. — 49-53.
38. Лукина Е. Гистиоцитозы как заболевания макрофагальной системы // Тер. архив. — 1996. — 7. — 82-88.
39. Махаонова Л., Дурнов Л. Гистиоцитарные заболевания у детей. — М.: Медицинское информационное агентство, 2004. — 103 с.
40. Махонова Л., Морозова О., Попа А. Современные проблемы терапии гистиоцитоза из клеток Лангерганса у детей (обзор литературы) // Детская онкология. — 2005. — 4. — 41-48.
41. Минков М. Лечение незлокачественных гистиоцитозов у детей: Автореф. дис... д-ра мед. наук. — М., 2005.
42. Минков М., Гаднер Х. Гистиоцитоз из клеток Лангерганса: результаты кооперированных исследований // Вопр. гематологии/онкологии и иммунопатологии в педиатрии. — 2004. — 3. — 7-10.
43. Минков М., Новичкова Г., Цельгер Г. и др. Гистиоцитозы детского возраста. — Москва — Вена: Макс-Пресс, 2005. — 30-36.
44. Солопова Г., Байдильдина Д., Жарикова Л. и др. Применение 2-хлордезоксиаденозида в терапии гистиоцитоза из клеток Лангерганса у детей // Онкогематология. — 2010. — 3. — 8-15.