
Газета «Новости медицины и фармации» Кардиология и ревматология (536) 2015 (тематический номер)
Вернуться к номеру
Дефіцит субкласів IgG: клініка, діагностика, лікування
Авторы: Мальцев Д.В. — Центр клінічної імунології та алергології Інституту експериментальної і клінічної медицини НМУ імені О.О. Богомольця, м. Київ
Разделы: Справочник специалиста
Версия для печати
Статья опубликована на с. 15-32
Вступ
Дефіцит субкласів IgG — група поширених у популяції імунодефіцитних хвороб, пов’язаних зі зниженням сироваткової концентрації одного або кількох різновидів імуноглобулінів класу G [137]. Хоча частота цих порушень у світі достеменно не відома, дефіцит субкласів IgG вважається однією з найпоширеніших імунних дисфункцій. Марек Якобисяк засвідчує, що дефіцит одного з субкласів IgG має місце принаймні у 25 % випадків серед дітей, які перенесли щонайменше 7 інфекційних епізодів за 1 рік. Однак, за результатами нещодавнього рандомізованого дослідження, проведеного P. Wågström зі співавт., частота інфекційних епізодів є недостатнім за валідністю критерієм дефіциту субкласів IgG, оскільки також має значення тяжкість, атиповість і пролонгованість інфекцій, а також розвиток автоімунних, алергічних і неопластичних ускладнень [176].
Виділяють 4 субкласи IgG, що позначаються арабськими цифрами в порядку зменшення їх вмісту в сироватці крові (IgG1, IgG2, IgG3, IgG4) (рис. 1).
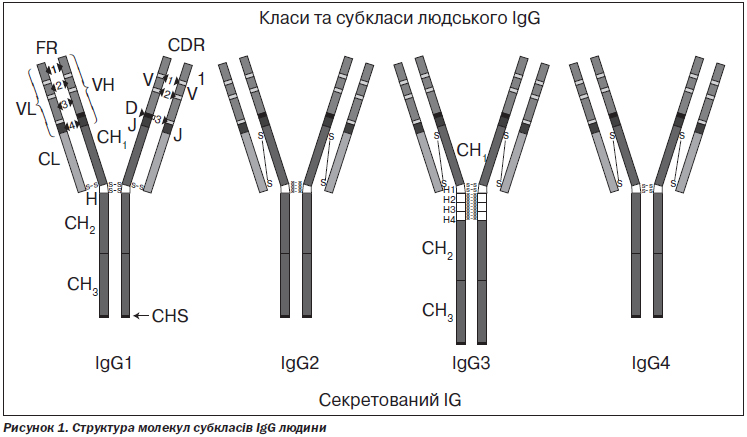
Субкласи різняться структурою константних ділянок, специфічністю до антигенів та функціональною активністю (табл. 1). Клінічна картина дефіциту різних субкласів IgG дещо відрізняється відповідно до їх функціонального призначення.

Якщо говорити про послідовність синтезу різних субкласів IgG під час імунної відповіді, то згідно з останньою моделлю, запропонованою A.M. Collins, K.J.L. Jackson, при надходженні антигену спочатку виробляються низькоафінні IgE та IgG3, що є складовими негайної імунної відповіді, потім — високоафінні IgG1 та IgG2, що забезпечують кліренс середовищ від антигену, і зрештою — IgG4, що нарощують специфічність відповіді і реалізують блокуючий ефект щодо прозапальної активності інших імуноглобулінів [27].
Етіологія
Добре відомі сімейні випадки дефіциту субкласів IgG вказують на генетичну основу цих розладів [31, 54, 102, 162]. V.A. Oxelius зі співавт. вивчили вміст субкласів IgG у матерів імуноскомпрометованих немовлят зі стрептококовою септицемією. У 13 з 19 матерів відзначався аналогічний імунодефіцит: 10 із них мали низьку концентрацію IgG2, 9 — IgG1, а 4 — IgG3. Відзначалася вірогідна різниця з контрольною групою здорових осіб [125]. Ці непрямі дані спонукали до проведення поглиблених генетичних досліджень при ізольованому дефіциті субкласів, завдяки яким встановили, що гомозиготні делеції генів константних ділянок імуноглобулінів 14-ї хромосоми є класичною причиною розвитку цього імунодефіциту у людей [90]. Так, T. Terada зі співавт. виявили делеції генів C alpha 1, psi C gamma, C gamma 2, C gamma 4 і C epsilon у пацієнта з дефіцитом IgG2, IgG4, IgA та IgE [164]. Тим не менше H. Tashita зі співавт. ідентифікували інсерцію 1793insG в 4-му екзоні C gamma 2 у двох пацієнтів з ізольованим дефіцитом IgG2 [163]. Відомі транслокації генів субкласів IgG, що тісно пов’язані з непластичними ускладненнями [77]. Поліморфізми генів константних ділянок імуноглобулінів також можуть обумовлювати розвиток первинного імунодефіциту [117] (рис. 2).
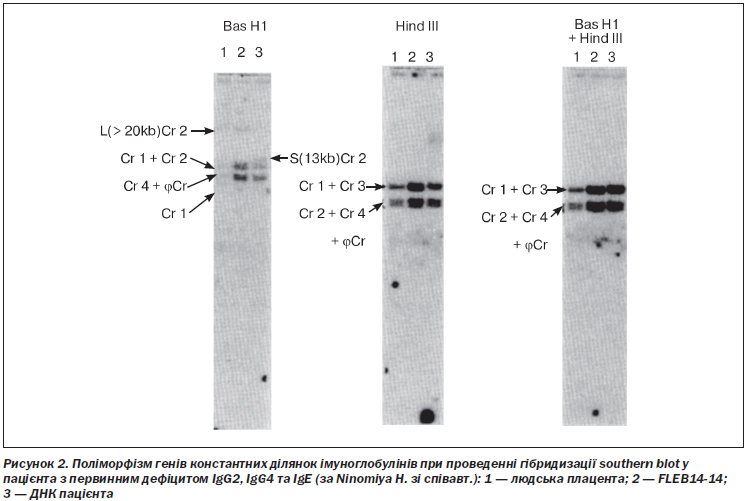
Зокрема, алотип G3(g) є причиною ізольованого дефіциту IgG3 у людей, причому він часто поєднується з алотипом G2(n), що обумовлює комбінування з прихованим дефіцитом IgG2 [96]. У зв’язку з цим Y. Miwa зі співавт. не виявили делецій у структурних генах константної ділянки важких ланцюгів імуноглобулінів при сімейних формах дефіциту субкласів IgG, що вказувало на іншу генетичну природу хвороби [111].
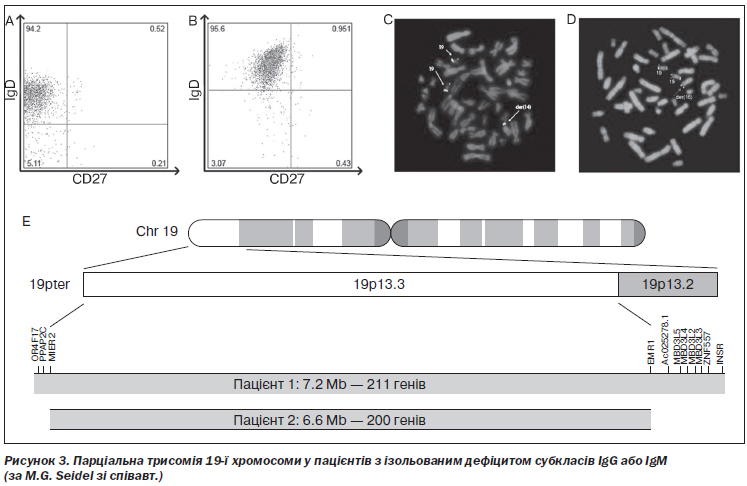
M.G. Seidel зі співавт. показали, що парціальна трисомія 19p13 призводила до комбінованого дефіциту IgG1 і IgG3 в одного пацієнта та вибіркового дефіциту IgМ — в іншого [148] (рис. 3). Y. Zhao зі співавт. описали ізольований дефіцит IgG2 у пацієнта з точковою мутацією гена C gamma 2, що призводила до аномального сплайсингу [180]. R. Gallina зі співавт. у дослідженні за участю пацієнтів із верифікованим дефіцитом IgG4 та їхніх найближчих родичів показали тісну асоціацію цього імунодефіциту і структурного поліморфізму гена IGHC. Експресія алелі HLA-D сприяла розвиткові дефіциту IgG4 в таких випадках [46].
Дефіцит субкласів IgG може бути генетично детермінованим імунорегуляторним розладом, пов’язаним з утрудненням процесу переключення ізотипів антитіл під час імунної відповіді. Так, S. Péron зі співавт. описали гомозиготну мутацію гена PMS2, що призводила до порушення переключення класів імуноглобулінів під час імунної відповіді й була причиною ізольованих дефіцитів класів та субкласів антитіл [132]. H. Kaneko зі співавт. виявили в пацієнтів із вибірковим дефіцитом IgG транскрипційний розлад у гамма-регіоні переключення ізотипів та гені IL-4 [73]. Відповідно до цього аналіз northern blot виявив порушення механізму відкриття хроматинової структури в зоні переключення ізотипів константних ділянок важких ланцюгів імуноглобулінів при дефіциті IgA та субкласів IgG [84].
Дефіцит субкласів IgG може бути наслідком субкомпенсації транзиторної гіпоімуноглобулінемії немовлят. R. Nettagul зі співавт. описали поступову зміну фенотипу транзиторної гіпоімуноглобулінемії немовлят, що проявлялася хронічною діареєю та 5 епізодами сепсису, до ізольованого дефіциту IgG2 з пом’якшенням клінічної картини і розвитком рецидивного середнього отиту і пневмоній [115]. Дефіцит субкласів IgG може бути проявом субкомпенсованого загального варіабельного імунодефіциту. Так, A. Aghamohammadi зі співавт. встановили, що у родичів пацієнтів із загальним варіабельним імунодефіцитом у 30 % випадків реєструється фенотип дефіцитів класів імуноглобулінів або субкласів IgG, здебільшого IgG2 та IgG4 [3].
C. Cunningham-Rundles зі співавт. встановили, що алель HLA B38 асоційована з дефіцитом субкласів IgG, особливо в разі поєднання цього розладу з дефіцитом IgА [28]. Алелі A*01-B*08, A*02-B*44 та A*29-B*44 тісно пов’язані як із дефіцитом субкласів IgG, так і загальним варіабельним імунодефіцитом, що вказує на спільність походження цих розладів [10]. Куріння може поглиблювати наявний дефіцит субкласів IgG, як це показали в спеціальному дослідженні V. Popa зі співавт. [135].
Епідеміологія
За даними T.W. Kuijpers, у дітей, які страждають від рецидивних респіраторних інфекцій, дефіцит субкласів IgG зустрічається у 20 % випадків [86], однак поширеність імунодефіциту в загальній популяції залишається неуточненою. Як повідомляють R. Gallina зі співавт., серед італійців ізольований дефіцит IgG4 зустрічається з частотою 1 випадок на 400 мешканців [46].
У дітей дефіцит субкласів IgG зустрічається частіше, ніж у дорослих, хоча поширеність імунодефіциту серед дорослого населення також є високою. Вважають, що чоловіки і жінки хворіють з однаковою частотою. Однак L.A. Hanson зі співавт. встановили, що серед дітей від дефіциту субкласів IgG частіше страждають хлопчики (3 хлопчики/1 дівчинка), тоді як серед дорослих імунодефіцит частіше реєструється у жінок (1 чоловік/3 жінки) [59]. C. Feldman зі співавт. у спеціально спланованому дослідженні не виявили суттєвих відмінностей у клінічних проявах імунодефіциту серед представників білої і чорної раси [43].
V. Popa зі співавт. виявили гуморальний імунодефіцит у 58 із 136 дорослих пацієнтів із рецидивними респіраторними інфекціями. Дефіцит IgA відзначався в 3 випадках, дефіцит IgM — також у 3, тоді як 52 особи мали дефіцит загального IgG або субкласів IgG. Найчастіше зустрічався ізольований дефіцит певного субкласу IgG, особливо IgG3 або IgG4, а також м’яке зниження концентрації загального IgG (між 450 і 650 мг/дл) [136].
E. Stanford зі співавт. вивчили поширеність дефіциту субкласів серед 172 дітей з інвазивною пневмококовою інфекцією. Гуморальні імунні порушення виявлені в 11 % випадків. Відзначалася кореляція між глибиною дефіциту IgG та кількістю серотипів пневмокока, до яких формувалася непротективна імунна відповідь після вакцинації. Невдачі вакцинації були вдвічі частіше асоційовані з дефіцитом IgG, ніж у загальній групі. Було багато випадків субнормальних рівнів IgG або транзиторних порушень, які, однак, призводили до розвитку тяжких клінічних симптомів [155].
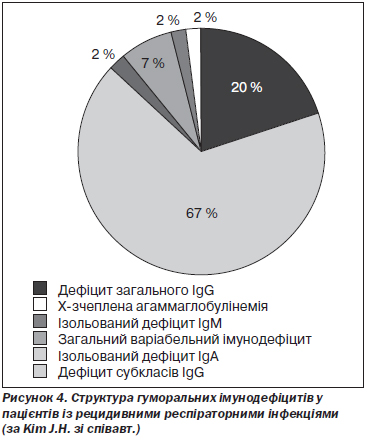
J.H. Kim зі співавт. провели ретроспективне дослідження за участю 55 дорослих із первинними імунодефіцитами, діагноз яких був встановлений у період між 1998 і 2009 роком у територіальному медичному центрі Кореї. Дефіцити субкласів IgG зустрічалися найчастіше і мали місце в 67 % випадків. Дефіцит загального IgG діагностовано у 20 %, IgМ — у 7 %, загальний варіабельний імунодефіцит — у 2 % випадків, а Х-зчеплена агаммаглобулінемія — ще у 2 % випадків. Дефіцити IgG3 та IgG4 зустрічалися найчастіше [80] (рис. 4).
M.L. Santaella зі співавт. встановили, що дефіцит субкласів IgG зустрічається щонайменше у 18,5 % випадків серед пацієнтів із дефіцитом IgА, тоді як дефіцит манозозв’язуючого білка — лише у 3,7 % випадків. Додатковий субкласовий дефіцит збільшував тяжкість інфекції та підвищував ризик розвитку атопії [145]. У той самий час компенсаторна гіпергаммаглобулінемія зустрічається щонайменше в 60 % випадків при дефіциті IgА [97].
M. Vendrell зі співавт. показали, що дефіцит субкласів IgG відзначався щонайменше в 11 % випадків серед пацієнтів із бронхоектазами невідомого походження. Особливо часто мав місце дефіцит IgG2, що обумовлював аномально слабку відповідь до Streptococcus pneumoniae і H.influenzae type b. Імовірність дефіциту IgG2 становила 58 %, якщо в клінічній картині паралельно відзначався рецидивний середній отит [171].
Y.S. Thakar зі співавт. у контрольованому дослідженні за участю 734 пацієнтів з інфекційним синдромом виявили дефіцит субкласів IgG у 38 випадках (5 %), що було вірогідно вище, ніж серед 100 здорових осіб контрольної групи [166].
M. Martinot зі співавт. дослідили поширеність дефіциту класів антитіл серед 119 пацієнтів з інвазивними інфекціями, викликаними Streptococcus pneumoniae і Haemophilus influenzae. У 18 випадках відзначалися менінгіти, у 79 — пневмонії, у 22 — інші тяжкі ураження. Гуморальні імунні порушення відзначалися у 37 із 119 пацієнтів (31 % випадків). Серед них у 37,8 % випадків мав місце дефіцит IgG1 [105].
A. Stead зі співавт., вивчаючи дані пацієнтів із множинними бронхоектазами, виявили ізольований дефіцит загального IgG у 3 із 56 хворих. Ще у 13 осіб мав місце дефіцит субкласів IgG, причому ізольований дефіцит IgG4 був найчастішим (9 випадків). Вакцинація демонструвала неможливість вироблення протективного титру антипневмококових антитіл в 1 із 29 випадків, при яких мав місце низький базальний рівень антиполісахаридних антитіл [156].
T. Ovesen зі співавт. виявили ізольовані гуморальні імунодефіцити у 5 із 18 дітей із хронічною посттимпанічною отореєю. Зокрема, тотальний і парціальний дефіцит IgА, дефіцит IgG та дефіцит манозозв’язуючого білка. Ще у 8 пацієнтів гуморальні порушення комбінувалися з дефіцитом цитотоксичних Т-лімфоцитів або природних кілерів [123].
N.E. Karaca зі співавт. показали, що серед дітей із дефіцитами субкласів IgG ізольований дефіцит IgG3 трапляється в 77 % випадків, комбінований дефіцит IgG2 та IgG3 — у 14 %, а ізольований дефіцит IgG2 — у 9 % випадків. Основними клінічними проявами були рецидивні інфекції верхніх дихальних шляхів, пневмонії, гастроентерит та інфекції сечовивідних шляхів. Атопія розвивалася в 15 % випадків [74].
N. Kutukculer зі співавт. спостерігали 87 дітей із дефіцитом класів імуноглобулінів і субкласів IgG. Найчастішим варіантом імунодефіциту був парціальний дефіцит IgA в комбінації з дефіцитом IgG3 (41 %). Також діагностували ізольований парціальний дефіцит IgA (32 %), вибірковий тотальний дефіцит IgA (8 %), парціальний дефіцит IgA у комбінації з дефіцитом субкласів IgG2-G4 (6 %) та ізольований дефіцит субкласів IgG (13 %). Показано, що траплялися як транзиторні порушення, що зазнавали спонтанної компенсації на тлі профілактичної антибіотикотерапїї (50–60 %) протягом 50–60 місяців, так і прогресуючі форми імунодефіциту, що погіршувалися з часом (30–40 % випадків) [87].
Класифікація
При ізольованому дефіциті субкласу IgG відзначається зниження сироваткової концентрації одного з різновидів цього імуноглобуліну. Іноді трапляються комбіновані порушення субкласів IgG, здебільшого — дуальні. Найчастішою комбінацією є дефіцит IgG2 та IgG4, хоча можливі найрізноманітніші поєднання. Відомі також поєднані гуморальні порушення, при яких дефіцит одного чи кількох субкласів IgG асоційований із низькою концентрацією інших класів імуноглобулінів, здебільшого — IgА, хоча існують повідомлення про зв’язок із дефіцитом IgМ, IgЕ або IgD. Так, Y. Miwa зі співавт. описали в одного пацієнта комбінований дефіцит IgG2 та IgG4, у другого — поєднане порушення, що включало дефіцит IgG2, IgG4 та IgА, а ще у двох — поєднання дефіцитів IgG2 та IgА [111]. При більш глибоких порушеннях зазвичай говорять про дизімуноглобулінемію. При одночасному дефіциті IgG, IgА, IgЕ і IgD діагностують гіпоімуноглобулінемію (< 7 г/л), а при зниженні сироваткового вмісту всіх класів імуноглобулінів — пангіпоімуноглобулінемію [166]. Часом дефіцит субкласів IgG супроводжується компенсаторною гіперімуноглобулінемією, що може зменшувати прояви інфекційного синдрому, однак асоційована з вищим ризиком імунозапальних, автоімунних та алергічних ускладнень [172].
За тяжкістю можна розрізняти тотальні і парціальні дефіцити субкласів IgG, однак надійних критеріїв їх чіткого розмежування не запропоновано. N. Visitsunthorn зі співавт. пропонують діагностувати парціальний дефіцит при зниженні сироваткової концентрації субкласу більше ніж на 2 стандартних відхилення від нижньої межі норми або на підставі аномально низького рівня співвідношення вмісту конкретного субкласу до загального IgG. При цьому автори діагностували парціальний дефіцит субкласів на підставі аномального співвідношення у 89,3 % випадків, і лише у 12,7 % — за низькою сироватковою концентрацією [175].
Можливі стійке і транзиторне зниження сироваткової концентрації субкласів IgG при первинних імунодефіцитах. В обох клінічних ситуаціях описаний розвиток тяжких симптомів. Так, S. Ohga зі співавт. описали рецидивний пневмококовий менінгіт, гепатит і гнійний артрит у дитини з транзиторним комбінованим дефіцитом IgG2 та IgG4 [118].
Імунодефіцит може бути первинним (генетично детермінованим) та вторинним (набутим). Винятком є випадки транслокацій генів імуноглобулінів, викликані вірусом Епштейна — Барр, наприклад у разі лімфоми Беркітта, при яких формується генетично детермінований набутий дефіцит субкласів IgG [192]. Вторинний дефіцит може бути проявом іншої хвороби. Так, дефіцит тотального IgG зустрічається при міотонічній дистрофії [143], дефіцит IgG2 — при серпоподібноклітинній анемії [114], а дефіцит IgG4 — при синдромі Rothmund-Thomson [85]. Різні варіанти дефіциту субкласів можуть відзначатися при синдромі Кабукі [61] та гемохроматозі, обумовленому гомозиготною мутацією HFE C282Y [9].
Крім того, дефіцит субкласів може бути компонентом іншого імунодефіциту з ширшим фенотипом, наприклад синдрому Луї — Барр або дефіциту молекули CD19 [174]. Однак можливі випадкові комбінації незалежних імунних дисфункцій, особливо якщо ті часто зустрічаються в популяції. Зокрема, F. Resman зі співавт. описали некротичний міозит і септичний шок, викликаний Haemophilus influenzae type f, у пацієнта з дефіцитом IgG3 та манозозв’язуючого протеїну [141].
Іноді до вторинного імунодефіциту призводять антиконвульсанти. Так, карбамазепін може викликати вторинний дефіцит IgG1 і IgG2 [50], фенітоїн — IgG2 та IgG4 [65], а зонізамід — дефіцит IgG2 та IgA [103]. Крім того, протизапальний препарат сульфасалазин може зумовлювати поєднаний дефіцит IgG2 та IgA [91].
Дефіцит IgG2 часом розвивається внаслідок спленектомії, однак A.F. Kavanaugh, D.P. Huston описали парадоксальне усунення передіснуючого дефіциту IgG2 після видалення селезінки з приводу портальної гіпертензії і гіперспленізму [76]. Спленектомія може сприяти декомпенсації прихованого дефіциту субкласів [41]. A. Scoular зі співавт. описали дефіцит IgG2 і пов’язану з цим бактеріємію, викликану Fusobacterium nucleatum, у ВІЛ-інфікованого пацієнта з нормальною кількістю CD4+ Т-лімфоцитів [147]. Вторинний дефіцит субкласів IgG розвивається при хронічному лімфолейкозі, визначаючи розвиток бактеріальних ускладнень при цьому новоутворенні [44], однак слід враховувати і зворотний зв’язок — розвиток лімфопроліферативних пухлин у пацієнтів із первинним дефіцитом субкласів IgG [77]. Вважають, що перитонеальний діаліз може спричиняти вторинний дефіцит субкласів IgG [146], однак результати нещодавнього дослідження встановили, що саме дефіцит IgG2 і призводив у більшості випадків до ниркової недостатності внаслідок розвитку інфекційних та автоімунних уражень, що зумовлювало потребу в діалізі [79].
Слід враховувати, що іноді ізольований дефіцит субкласу IgG може супроводжуватися прихованим дефіцитом специфічних антитіл різних класів. Так, D.A. Van Kessel зі співавт. виявили серед 24 пацієнтів із дефіцитом IgG1 щонайменше 9 випадків дефіциту продукції специфічних IgG2 або IgA у відповідь на пневмококову вакцину, хоча загальні сироваткові рівні цих імуноглобулінів були нормальними [168]. Більше того, C. Lacombe зі співавт. описали 3 випадки трансформації ізольованого дефіциту IgG1 в дефіцит IgG2 [88].
Клінічні прояви
Дефіцит субкласів IgG обумовлює розвиток клінічних симптомів інфекційних, алергічних, автоімунних і неопластичних уражень у людей. Загалом тяжкість стану пацієнтів різниться в разі поєднаних, комбінованих та ізольованих порушень, однак ці відмінності можуть не справджуватися в конкретних випадках.
Поєднані форми. Якщо дефіцит субкласів IgG поєднується з парціальним або тотальним дефіцитом IgA, то зазвичай формуються більш тяжкі клінічні прояви, ніж при ізольованому дефіциті субкласів [29]. Так, N. Kutukculer зі співавт. виявили такі клінічні прояви поєднаних форм імунодефіциту: рецидивні інфекції верхніх дихальних шляхів (76 %), пневмонія (14 %), гострий гастроентерит (3 %), інфекції сечовивідних шляхів (3 %), синусит (2 %) і гострий середній отит (2 %). Атопія траплялася у 24 % випадків (n = 87) [87].
F. DeBaets зі співавт. виявили гуморальні імунодефіцит серед дітей із хронічним бронхітом у 85 % випадків. 9 пацієнтів (17 %) мали дефіцит IgG2, 9 (17 %) — IgG3, а 20 осіб (38 %) — дефіцит IgG4. Ще 9 дітей (17 %) страждали від дефіциту IgA, а 8 (15 %) — від комбінованого дефіциту субкласів IgG. Відзначалася вірогідна різниця порівняно з контрольною групою, яку становили здорові діти. Виявлена кореляція між глибиною імунодефіциту й тяжкістю інфекційних проявів [33].
T. Saleem зі співавт. описали дефіцит IgА, IgG2 і IgG4 у 17-річної пацієнтки з анамнезом рецидивних бактеріальних інфекцій, включаючи туберкульоз. Численні курси антибіотиків не зменшили кількості рецидивів інфекції. Найчастішими проявами були: пневмонія, гастроентерит та максилярний синусит. Чотири рази вона успішно приймала в/в імуноглобулін при загостренні стану, однак тривалої імуноглобулінотерапії не проводили. Через 5 років у неї розвинулася неходжкінська лімфома [144].
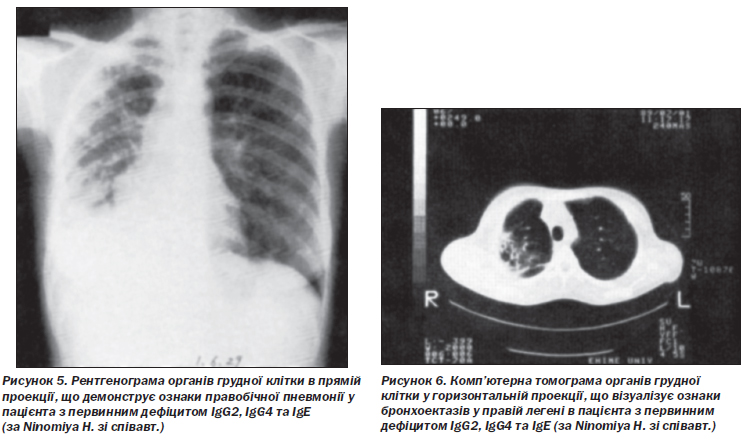
G. Ideura зі співавт. описали дефіцит IgМ та IgG4 у 37-річної пацієнтки з рецидивною бронхопневмонією та фіброепітеліальним поліпом у бронху. Згодом у неї розвинулася алергічна системна еритема у відповідь на антибактеріальні препарати. Відзначалися зниження кількості CD20+ В-лімфоцитів та відсутність В-клітин пам’яті [62]. Натомість K. Pengsaa зі співавт. повідомили про хронічний лямбліозний ентерит у 15-річного пацієнта з дефіцитом IgМ та IgG4 [131]. H. Ninomiya зі співавт. повідомили про рецидивні пневмонії (рис. 5) та множинні бронхоектази легень (рис. 6) у пацієнта з поєднаним дефіцитом IgG2, IgG4 та IgЕ, зумовленим поліморфізмом генів константних ділянок імуноглобулінів.
Комбіновані порушення. Одночасний дефіцит кількох субкласів IgG призводить до дещо слабших клінічних проявів порівняно з поєднаними порушеннями, які, однак, зазвичай тяжчі, ніж при ізольованих дефіцитах. Слід враховувати, що тяжкість симптомів обумовлена не тільки глибиною імунодефіциту, але й реалізацією компенсаторних механізмів. J.De Gracia зі співавт. виявили дефіцит субкласів серед пацієнтів із множинними бронхоектазами в 48 % випадків: у 19 — дефіцит IgG2, 2 — дефіцит IgG3, 3 — дефіцит IgG4 і в 7 — комбіновані порушення. При цьому у всіх осіб відзначалося зростання сироваткової концентрації загального IgG, IgG1 і IgA [30]. T. Klingebiel зі співавт. описали пневмонію, викликану Chlamydia trachomatis, у пацієнта з дефіцитом субкласів IgG2 та IgG4. Ускладненням був пневмоторакс, що потребував виконання хірургічного втручання. Дитина також страждала від імпетиго [82].
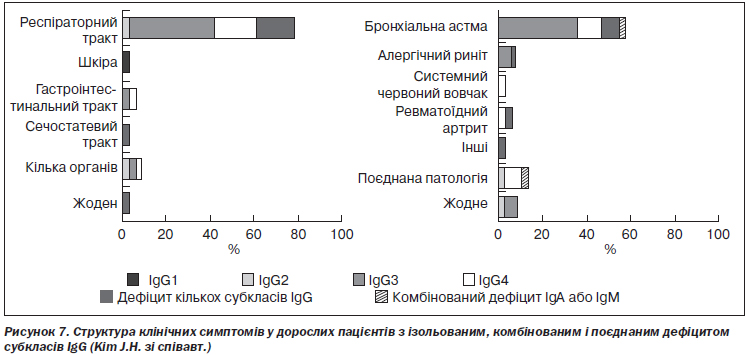
Ізольовані форми дефіциту. У таких випадках частіше відзначаються асимптомні варіанти перебігу хвороби, ніж у разі поєднаних і комбінованих порушень, оскільки має місце менша за глибиною імунна недостатність і ширші можливості для компенсації імунодефіциту. J.H. Kim зі співавт. вивчили структуру клінічних проявів ізольованого дефіциту субкласів IgG у дітей. Виявилось, що бактеріальні інфекції верхніх і нижніх дихальних шляхів зустрічалися в 76 % випадків, а інфекції множинної локалізації, включаючи ураження сечовивідних шляхів і коліт, — в 11 % випадків. Бронхіальна астма, алергічний риніт і декілька автоімунних хвороб також зустрічалися серед цих пацієнтів [80] (рис. 7).
C. Feldman зі співавт. у спеціально спланованому дослідженні показали асоціацію ізольованого дефіциту субкласів IgG із рецидивними респіраторними інфекціями, атопією та бронхоектазами у дорослих пацієнтів [43]. J.P. Faller зі співавт. продемонстрували асоціацію дефіциту субкласів IgG і фульмінантної форми менінгококової пурпури [40].
J.C. Barton зі співавт. у спеціально спланованому дослідженні за участю 212 дорослих показали тісну асоціацію дефіциту субкласів IgG і оперізуючого герпесу. У таких пацієнтів аномально часто відзначалися алелі HLA-A*01 та B*08. Протягом 7,5 року спостереження частота рецидивів оперізуючого герпесу в пацієнтів із дефіцитом субкласів IgG була в 5 разів вища, ніж в імунокомпетентних осіб [8, 12].
V. Popa продемонстрував зв’язок дефіциту субкласів IgG, так само як і загального варіабельного імунодефіциту, з епізодами обструкції дихальних шляхів у дорослих, що розвиваються на тлі рецидивних респіраторних інфекцій (n = 42) [135].
Загальний IgG
Під дефіцитом загального IgG розуміють паралельне зниження сироваткової концентрації всіх субкласів за умови нормального вмісту інших ізотипів імуноглобулінів. Може бути субкомпенсованою формою загального варіабельного імунодефіциту. Зумовлює неінформативність серологічних тестів при діагностиці інфекцій. S.L. Johnston зі співавт. описали редицивні шкірні й пульмональні інфекції, нефротичний синдром, цукровий діабет 1-го типу в дорослого пацієнта з тотальним дефіцитом IgG. У сечі не відзначалося молекул IgG, тому втрата IgG через нирки була виключена. Досягнута компенсація клінічного стану під впливом в/в імуноглобуліну [67]. A. Pardillos Tomé зі співавт. описали класичну саркому Капоші, викликану вірусом герпесу 8-го типу, у пацієнта з ізольованим дефіцитом загального IgG [128].
Дефіцит IgG1
Цей субклас розпізнає переважно білкові антигени і становить основу пулу загального IgG у сироватці крові людини. Клінічні прояви ізольованого дефіциту IgG1 можуть нагадувати загальний варіабельний імунодефіцит. Зазвичай відзначається зниження рівня загального IgG у сироватці крові, тому цей імунодефіцит можні виявити в рутинній імунограмі. Частіше за інші дефіцити субкласів поєднується з дефіцитами інших ізотипів антитіл. Деякі автори вважають дефіцит IgG1 різновидом загального варіабельного імунодефіциту [112].
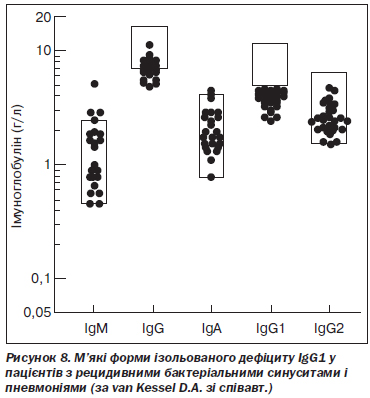
C. Lacombe зі співавт. верифікували дефіцит IgG1 у 119 випадках серед 3005 пацієнтів. Клінічні симптоми у вигляді рецидивних бактеріальних інфекцій відзначалися у 83,2 % випадків. Також реєструвалися атопічний дерматит, природжені вади серця та автоімунні розлади, однак найчастіше виявляли бронхіальну астму (кожен п’ятий пацієнт). У більшості випадків відзначалися сімейні форми імунодефіциту [88]. D.A. Van Kessel зі співавт. повідомили про 24 випадки м’якого ізольованого дефіциту IgG1. Основними клінічними проявами були рецидивні бактеріальні синусити і пневмонії [168] (рис. 8).
K. Kallio-Laine зі співавт. у спеціально спланованому дослідженні продемонстрували, що сироваткова концентрація IgG1 була нижчою (p = 0,009), а частота виявлення ізольованого дефіциту IgG1 більшою (p < 0,001) у пацієнтів із рецидивним менінгітом Молларе, викликаним вірусом простого герпесу 2-го типу порівняно з контрольною групою. Ризик нового епізоду менінгіту зростав зі зниженням концентрації IgG1 (показник інциденту 2,05). Експресія алелей HLA-DRB1*01 і B*27 була асоційована з дефіцитом IgG1 і високим ризиком нейроінфекції [70].
J. Hassett зі співавт. описали рецидивний ентероколіт, викликаний Clostridium difficile, у пацієнта з дефіцитом IgG1 [60].
IgG2
Ці антитіла виробляються проти полісахаридних антигенів, тому в пацієнтів із дефіцитом IgG2 знижується резистентність передовсім до Streptococcus pneumoniae та Haemophilus influenzae, що створює подібність із дефіцитом специфічних антитіл [64]. Це найчастіша форма дефіциту субкласів IgG у дітей. P.G. Shackelford зі співавт. виявили дефіцит IgG2 в 7 випадках серед 30 дітей із рецидивними бактеріальними інфекціями, включаючи бактеріальні синусити, пневмонії та пневмококовий менінгіт [150], хоча C.S. Beck і D.C. Heiner ідентифікували цю імунну дисфункцію лише у 4 із 422 пацієнтів із тяжкими синупульмональними інфекціями [15]. R. Inoue зі співавт. описали дефіцит IgG2 у поєднанні з дефіцитом продукції гамма-інтерферону [63]. C.L. Gordon зі співавт. у когортному дослідженні з використанням мультиваріативного аналізу показали тісну асоціацію тяжких форм грипу, викликаного штамом H1N1, з ізольованим дефіцитом IgG2 [51]. X. Escobar-Pérez зі співавт. описали 4 випадки бактеріального менінгоенцефаліту, викликаного Streptococcus pneumoniae та Haemophilus influenzae type b у пацієнтів з ізольованим дефіцитом IgG2. Показана користь від застосування в/в імуноглобуліну в таких випадках [38]. D.N. Gottsegen зі співавт. описали розвиток пневмококового остеомієліту [53], а J.L. Bass зі співавт. — рецидивної менінгококцемії у пацієнтів з ізольованим дефіцитом IgG2 [13].
IgG3
Цей субклас IgG проявляє специфічність до білкових антигенів, тому при його дефіциті знижується резистентність насамперед до Str.pyogenes, що експресує М-протеїн, та Moraxella catarrhalis. Це найчастіша форма дефіциту субкласів IgG серед дорослих. Зазвичай розвиваються хронічні стрептококовий тонзилофарингіт та бронхіт, викликаний моракселою. Також знижена резистентність до вірусних агентів, що містять багато білкових антигенів, що є особливістю імунодефіциту.
N. Visitsunthorn зі співавт. показали, що ізольований дефіцит IgG3 зустрічався у 56,4 % випадків серед тайських дітей із дефіцитами субкласів IgG. Клінічний дебют припадав на період від народження до 5 років. Основним клінічним проявом був рецидивний бактеріальний синусит, що траплявся у 83,6 % випадків [175].
V.A. Oxelius зі співавт., вивчаючи структуру дефіциту IgG3, діагностували ізольовану форму дефіциту IgG3 в 59,5 % випадках серед 313 пацієнтів. Крім того, у 36,5 % випадках мав місце комбінований дефіцит IgG1 та IgG3, а ще в 5 % — IgG2 та IgG3. Пацієнти з ізольованим дефіцитом IgG3 страждали від рецидивних інфекцій верхніх дихальних шляхів, бронхітів, бронхопневмоній і бронхіальної астми. Особи з комбінованим дефіцитом IgG1 та IgG3 частіше хворіли на хронічну обструктивну хворобу легень і пневмонії. Крім того, у пацієнтів з ізольованим дефіцитом IgG3 відзначалися непоодинокі випадки цукрового діабету 1-го типу, пурпури Шенлейна — Геноха, рецидивної інфекції, викликаної вірусом герпесу 1-го типу, і бешихи. Цікаво, що в деяких родичів пацієнтів із дефіцитом IgG3 відзначався загальний варіабельний імунодефіцит та дефіцит IgA, що вказує на спільність походження цих розладів [124].
A. Vehapoglu зі співавт. описали рецидивний бактеріальний менінгіт у 6-річного хлопчика з ізольованим дефіцитом IgG3 [170]. F. Abrahamian зі співавт. показали асоціацію ізольованого дефіциту IgG3 з рецидивними бактеріальними респіраторними інфекціями, алергічним ринітом і бронхіальною астмою [2]. J.A. Snowden зі співавт. повідомили про ізольований дефіцит IgG3 у 35-річної пацієнтки з рецидивними інфекціями верхніх дихальних шляхів, синуситами і генералізованою лімфаденопатією. Показана компенсація стану після призначення в/в імуноглобуліну [153]. M. Armenaka зі співавт. у контрольованому дослідженні виявили асоціацію дефіциту IgG3 з хронічним рефрактерним синуситом у людей [7].
H. Mitsui зі співавт. повідомили про розвиток хронічної активної інфекції, викликаної вірусом Епштейна — Барр, що проявлялася рецидивними оральними й генітальними виразками, некротичними папулами на обличчі, спалахами високої температури тіла та індурованою еритемою на шкірі тулуба. Показана користь від комбінованої терапії за допомогою в/в імуноглобуліну та альфа-інтерферону [110]. A. Linde зі співавт. описали серію випадків реактивованих інфекцій, викликаних вірусом Епштейна — Барр і вірусом герпесу 6-го типу, у пацієнтів із дефіцитом IgG3. Автори показали, що клінічно маніфестне ураження розвивається лише в тому разі, коли в контексті дефіциту загального IgG3 відзначається дефіцит специфічних IgG3 до вірусу. Паралельно з цим нерідко мав місце дефіцит IgG2 до полісахаридних антигенів при нормальному загальному пулі IgG2, що пояснює тісну асоціацію вірусу Епштейна — Барр зі стрептококовою інфекцією у деяких хворих [96]. J.A. Snowden зі співавт. описали рецидивний ентеровірусний серозний лімфоцитарний менінгіт у пацієнта з ізольованим дефіцитом IgG3 [152]. K. Kallio-Laine зі співавт. описали серію випадків часто рецидивуючого лабіального герпесу, викликаного вірусом простого герпесу 1-го типу, у пацієнтів з ізольованим дефіцитом IgG3 та комбінованим порушенням, що включало дефіцит IgG1 і IgG3 [71].
M. Maes зі співавт. повідомили про синдром хронічної втоми і транслокацію ендотоксинів грамнегативних бактерій через стінку кишечника у 13-річної дівчинки з вибірковим дефіцитом IgG3 [107]. J. Ambrozic зі співавт. повідомили про рецидивний бактеріальний сепсис та серонегативний олігоартрит у пацієнта з ізольованим IgG3 [5].
IgG4
Ізольований дефіцит цього субкласу часто має безсимптомний перебіг у зв’язку з малою нормальною концентрацією IgG4 в сироватці крові, що обумовлює широкі можливості для компенсації. Імуноглобулін має протизапальні властивості і в разі атопії виступає в ролі блокуючого антитіла. R.B. Moss зі співавт. виявили ізольований дефіцит IgG4 в 17 % випадків серед дітей із рецидивними респіраторними інфекціями, що становило разючий контраст із контрольною групою, яку становили здорові діти (n = 250). Додатковими клінічними проявами були атопічний дерматит, бронхіальна астма, рецидивний гастроентерит [113]. A. Rawat зі співавт. описали ізольований дефіцит IgG4 у дитини з бронхоектазами, обумовленими частими випадками бактеріальних бронхопульмональних інфекцій [139]. Раніше повідомляли про розвиток хронічної діареї і рецидивного лямбліозу при дефіциті цього субкласу. P. Pavone зі співавт. показали, що у пацієнтів із синдромом Дауна ішемічний інсульт асоційований із дефіцитом IgG4, що пов’язали зі зниженою резистентністю до Mycoplasma pneumoniaе і Streptococcus oralis [130].
O. Karaman зі співавт. виявили дефіцит субкласів у 31,6 % випадків серед немовлят зі свистячим диханням, причому переважно відзначався дефіцит IgG4 [75]. R. Castro зі співавт. описали тяжкий зовнішній отит і мастоїдит у дитини з ізольованим дефіцитом IgG4. Пацієнту виконано декілька мастоїдектомій і пластичних операцій із приводу рецидивів інфекційного ураження. Резидуальним дефектом було зниження слуху з іпсилатерального боку [25].
F. Levendoglu зі співавт. повідомили про спінальний епідуральний абсцес із прогресуючою параплегією у 16-річного пацієнта з ізольованим дефіцитом IgG4 [94].
Автоімунні прояви
У пацієнтів із дефіцитом субкласів IgG може розвиватися цукровий діабет 1-го типу і пурпура Шенлейна — Геноха [124], первинний біліарний цироз [161], автоімунна тромбоцитопенічна пурпура [164], тиреоїдит Хашимото і гемолітична анемія [47], комбіновані автоімунні цитопенії [41], синдром Евана [173]. Зокрема, M.S. Rahiminejad зі співавт. виявили дефіцит субкласів IgG в 11 % випадків серед пацієнтів з автоімунною тромбоцитопенічною пурпурою [138]. Про асоціацію з цим автоімунним розладом повідомляли також P.G. Shackelford зі співавт. [150]. Натомість P.M. Villiger зі співавт. описали синдром Евана у дорослого пацієнта, який страждав від комбінованого дефіциту субкласів IgG2 та IgG4 [173].
K. Suyama зі співавт. описали розвиток системного червоного вовчака в пацієнта з поєднаним дефіцитом IgA та IgG2, причому автоімунне ускладнення призвело до поглиблення імунної дисфункції до фенотипу загального варіабельного імунодефіциту [160]. A. Tamura зі співавт. повідомили про розвиток системного червоного вовчака в пацієнта з сімейним дефіцитом IgG2 і IgG4. У дитини розвинулася тампонада серця у зв’язку з ексудативним перикардитом [162]. N. Düzgün зі співавт. описали розвиток у 31-річної пацієнтки з дефіцитом загального IgG спочатку системного червоного вовчака, а потім — екстрапульмонального туберкульозу з ураженням шкіри, суглобів та менінгеальних оболонок мозку [36]. K. Kamei зі співавт. повідомили про 2 випадки автоімунного імунокомплексного ураження нирок у пацієнтів із дефіцитом субкласів IgG. У першому випадку мав місце дефіцит IgG2, IgG3, і IgG4. У нирках відзначалися депозити, представлені IgG1. Встановили діагноз мембранозної нефропатії. У другому випадку виявили дефіцит IgG2 та IgG4. Мали місце депозити із IgG1 та IgG3. Був встановлений діагноз мембранопроліферативного гломерулонефриту [72].
R.T. Hassani зі співавт. описали розвиток спочатку неспецифічного виразкового коліту, а потім — автоімунного васкуліту сітківки в пацієнта з ізольованим дефіцитом IgG1. Паралельно пацієнт страждав від рецидивних бактеріальних інфекцій [59].
A. Venuta зі співавт. повідомили про гіпергаммаглобулінемічну пурпуру у 12-річної пацієнтки з ізольованим дефіцитом IgG2 й анамнезом частих інфекційних епізодів [172]. P. Eriksson зі співавт. описали 6 випадків ізольованого дефіциту субкласів IgG, що призводив до розвитку синдрому Шегрена та гіпергаммаглобулінемічної пурпури [37]. Про подібний випадок повідомили L. Matter зі співавт. [106].
Нерідко розвиваються неврологічні автоімунні ускладнення. D.J. Likosky зі співавт. описали 3 випадки мультифокальної демієлінізуючої полінейропатії у пацієнтів з ізольованим дефіцитом IgG1 і/або IgG3 [95]. I. Meyts зі співавт. повідомили про оптичний нейромієліт у пацієнта з сімейним парціальним дефіцитом IgA та IgG3 [108]. L.P. Frohman зі співавт. діагностували рецидивний стероїдзалежний оптичний неврит у пацієнта з комбінованим дефіцитом IgG2 та IgG3 [45]. L.F. Bertoli зі співавт. виявили дефіцит субкласів IgG1 та IgG3 в 46,4 % випадків серед пацієнтів з автоімунною сенсоневральною приглухуватістю. Дефіцит IgG1 зустрічався в 10,7 %, дефіцит IgG3 — у 21,4 %, а комбіноване порушення — у 14,3 % випадків. Поширеність імунодефіциту була вірогідно вищою, ніж у контрольній групі, яку становили 275 здорових європейців. Поширеність можливої автоімунної сенсоневральної приглухуватості при субкласових дефіцитах становила 0,74 %. У 21,4 % випадків у таких пацієнтів відзначалися інші автоімунні розлади та був підвищеним вміст автоантитіл до колагену ІІ типу [17].
Як показали A. Jiménez зі співавт., ізольований дефіцит IgG2 може бути причиною синтезу автоантитіл до IgА, що призводить до розвитку вторинного дефіциту IgА. Також у таких пацієнтів підвищена частота синтезу антикардіоліпінових автоантитіл [66].
Алергічні прояви
У пацієнтів із дефіцитом субкласів IgG описані випадки бронхіальної астми [2], алергічного риніту [80], атопічного дерматиту [149], хронічного рефрактерного синуситу [169] та медикаментозної алергії [62]. Серед 62 пацієнтів із системними проявами атопії B. Bozkurt зі співавт. виявили дефіцит IgA, загального IgG, IgM та IgG3 у 12,9, 8, 6,5 і 1,6 % випадках відповідно, що вірогідно частіше, ніж у загальній популяції [20].
C. de Moraes Lui зі співавт. серед пацієнтів із бронхіальною астмою діагностували такі форми дефіциту субкласів IgG: дефіцит IgG3 (10/41), IgG4 (3/41), IgG2 (2/41), IgG1 (1/41), IgG3-IgG4 (4/41), IgG1-IgG3 (1/41), IgG1-IgG3-IgG4 (1/41). При цьому в пацієнтів із комбінацією астми й рецидивних респіраторних інфекцій переважав дефіцит IgG3, а серед осіб тільки з проявами астми — дефіцит IgG4 [32]. У деяких пацієнтів розвивається стероїдзалежна астма [39]. I.M. Outschoorn зі співавт. у контрольованому дослідженні показали асоціацію бронхіальної астми у дорослих із дефіцитом субкласів IgG2 і IgG3 [122]. B.G. Loftus зі співавт. виявили дефіцит субкласів IgG у 28 випадках серед 82 дітей із бронхіальною астмою на відміну від контрольної групи. Частіше за все зустрічався дефіцит IgG2. Глибина імунодефіциту корелювала зі швидкістю наростання сироваткової концентрації IgE [99].
S. O’Keeffe зі співавт. діагностували дефіцит субкласів IgG у 25,9 % випадків серед 58 пацієнтів із хронічною обструктивною хворобою легень. Найчастіше зустрічався ізольований дефіцит IgG2. Відзначалася вірогідна різниця з контрольною групою. Дефіцит субкласів був асоційований із поступовим погіршанням результатів спірометричних досліджень і виступав у ролі фактора прогресування обструктивної хвороби легень [120]. K. Lock зі співавт. діагностували дефіцит субкласів IgG у 26 із 99 пацієнтів із бронхіальною астмою і хронічною обструктивною хворобою легень. Глюкокортикостероїди поглиблювали наявний імунодефіцит, однак не були причиною його розвитку [98].
Tran N. Khai Hoan зі співавт. виявили дефіцит субкласів IgG у 13,7 % випадків серед пацієнтів із поліпами носової порожнини (161 особа) [167]. L. Vanlerberghe зі співавт. діагностували серед пацієнтів із хронічним рефрактерним риносинуїтом дефіцит субкласу IgG3 в 17,9 %, субкласу IgG2 — у 2 %, а комбіновані порушення — у 2,9 % випадків [169]. A. May зі співавт. виявили серед 220 пацієнтів із хронічними рефрактерним риносинуситом такі гуморальні імунні порушення: дефіцит загального IgG (3), IgG2 (n = 10), IgG1 (n = 6), IgG3 (n = 1) та IgG4 (n = 1) [107]. S. Chinratanapisit зі співавт. повідомили про одночасний розвиток рефрактерного риносинуситу й назальних поліпів у двох пацієнтів. У першого з них ідентифікували ізольований дефіцит IgG3, а у другого — комбіноване порушення, що включало дефіцит IgG2 та IgG3 [26].
A. Ojuawo зі співавт. діагностували дефіцит IgA, IgG2 і IgG4 в 43,3; 75,0 і 55,0 % випадків відповідно серед дітей з алергічним колітом і менше ніж у 5 % випадків у контрольній групі [119].
Онкологічні ускладнення
У пацієнтів із дефіцитом субкласів IgG розвиваються переважно мікроб-індуковані лімфопроліферативні пухлини [179]. T. Zenone зі співавт. описали 2 випадки лімфоми Ходжкіна у пацієнтів із дефіцитом субкласів IgG. Також повідомляли про розвиток неходжкінських лімфом і аденокарциноми шлунка [144], саркоми Капоші [128], лімфоми Беркітта [192]. Дефіцит IgG4, найімовірніше, відповідальний за розвиток остеосарком при синдромі Rothmund-Thomson [85]. Відомі описи лімфопроліферативних пухлин при транслокаціях і делеціях у генах субкласів IgG: гострого пре-В-лейкозу [77] і лімфоми, асоційованої зі слизовими оболонками [158], — при дефіциті IgG1, різних форм лейкозу — при дефіциті IgG2 [180], хвороби важких ланцюгів — при дефіциті IgG3 [4] та В-клітинної лімфоми — при дефіциті IgG4 [81].
Інші прояви
У дітей із дефіцитом субкласів можуть відзначатися ознаки затримки розвитку [151]. E. Pauly зі співавт. повідомили про розвиток синдрому нетримання пігменту в новонародженого з ізольованим дефіцитом субкласів IgG [129]. M. Hahn-Zoric зі співавт. показали, що пацієнти з дефіцитом IgG3 не формують протективної гуморальної імунної відповіді після імунізації кон’югованою вакциною, що містить капсулярний антиген Haemophilus influenzae type b і правцевий токсоїд [57]. У деяких пацієнтів із дефіцитом субкласів може розвиватися некласифікована ентеропатія [48]. L. Layward зі співавт. у контрольованому дослідженні показали асоціацію IgA-нефропатії і комбінованого дефіциту субкласів IgG1 та IgG4. Аномальне підвищення сироваткової концентрації IgA пояснили аберантною компенсацією дефіциту субкласів IgG [89].
Нерідко спостерігаються неврологічні ускладнення. Так, D. Wakefield зі співавт. продемонстрували асоціацію дефіциту субкласів IgG із синдромом хронічної втоми у людей [177]. Подібні результати отримали A.L. Komaroff зі співавт. [83] та R. Read зі співавт. [140] незалежно одні від одних. У дітей із дефіцитом субкласів можуть формуватися рефрактерні форми епілептичних синдромів, які позитивно реагують на введення в/в імуноглобуліну [133]. H. Caksen зі співавт. продемонстрували зв’язок дефіциту IgG4 і фебрильних судом у дітей [23]. Натомість C. Lenti зі співавт. у дослідженні сімей показали асоціацію ізольованого дефіциту IgG2 з фебрильними судомами в дітей [93]. M. Nieto зі співавт. виявили кореляцію між тяжкою міоклонічною епілепсією і дефіцитами IgA або IgG2 [116]. Натомість N.G. Bos-Veneman зі співавт. у контрольованому дослідженні показали асоціацію синдрому Туретта і дефіциту IgG3 [19].
Z. Mikhak зі співавт. повідомили про геміфаціальну мікросомію в сім’ї з гуморальним імунодефіцитом, обумовленим низькою сироватковою концентрацією IgG1, IgG2, IgA та специфічних антипневмококових антитіл [109]. V. Garg зі співавт. описали нодулярну лімфоїдну гіперплазію кишечника в пацієнта з дефіцитом IgG2 [47]. J.R. Farmer зі співавт. повідомили про розвиток пульмональної артеріовенозної мальформації в контексті рецидивних респіраторних інфекцій у пацієнта з поєднаним дефіцитом IgA, IgG2 та IgG4, у зв’язку з якою було виконано білатеральну трансплантацію легень [41]. Дефіцит субкласів зустрічається в 19,6 % випадків серед пацієнтів з інтерстиціальною хворобою легень, за даними дослідження V. Popa зі співавт. (n = 148) [136].
Клінічний перебіг
У деяких пацієнтів може мати місце безсимптомний перебіг, принаймні протягом певних проміжків часу. Компенсація імунодефіциту може здійснюватися різними шляхами: за рахунок гіпергаммаглобулінемії [151], посиленого вироблення інших ізотипів антитіл, наприклад IgA [89] або IgE [100], і навіть шляхом посиленої продукції інших, неуражених субкласів IgG. Так, L. Layward зі співавт. показали, що в пацієнтів із дефіцитом IgG1 відзначається атипове компенсаторне вироблення IgG2 до білкових антигенів тетанічного токсоїду [89]. A. Depiero зі співавт. продемонстрували, що посилене вироблення IgG3 компенсувало дефіцит IgG1, IgG2, IgG4 і IgA1 у пацієнта з великою делецією генів важких ланцюгів імуноглобулінів [35]. За даними A. Plebani зі співавт., підвищена продукція IgG1 та IgG3 компенсувала поєднаний дефіцит IgA1, IgG2, IgG4 та IgE у пацієнта з гомозиготною делецією генів константних ділянок імуноглобулінів A1-E [134]. Оскільки в таких випадках відмічається сурогатне заміщення функції втраченого субкласу, така компенсація не завжди досягає ефекту, не усуваючи прояви інфекційного синдрому, а часом призводить до розвитку імунозалежних побічних ефектів. Так, гіперпродукція загального IgG обумовлює розвиток гіпергаммаглобулінемічної пурпури та автоімунних ускладнень [172], гіперпродукція IgE призводить до алергічних уражень, включаючи бронхіальну астму [32], а посилене вироблення IgA або IgM — до так званих IgA- [89] та IgM-нефропатій [126] відповідно.
Кореляція між глибиною дефіциту субкласів і тяжкістю клінічних симптомів виражена слабше, ніж при гіпогаммаглобулінемії [112]. Навіть субнормальні рівні субкласів можуть бути асоційовані з тяжкими клінічними проявами [155]. Іноді дефіцит субкласів IgG зазнає декомпенсації з розвитком фенотипу загального варіабельного імунодефіциту [6]. Часом дефіцит субкласів зазнає тимчасової псевдокомпенсації під впливом тяжкої інфекції, найбільш імовірно, за рахунок її потенціюючої дії на імунну відповідь [69].
N.E. Karaca зі співавт. продемонстрували, що дефіцит IgG3 зазнавав спонтанної компенсації у 30 % випадків, а комбінований дефіцит IgG2 та IgG3 — у 35,7 % випадків протягом 6 років. Однак ізольований дефіцит IgG2 не зазнавав компенсації протягом усього періоду спостереження [74].
W. Carvalho Neves Forte зі співавт. описали еволюцію ізольованого дефіциту IgA до поєднаного порушення з дефіцитом IgG2 та IgG4 в одному випадку і до фенотипу загального варіабельного імунодефіциту — в іншому. В обох випадках показана очевидна користь від застосування в/в імуноглобуліну [24].
Діагностика
S. Jolles зі співавт. у спеціально спланованому дослідженні показали, що визначення калькульованого глобуліну (< 18 г/л) може бути скринінговим тестом при діагностиці гуморальних імуноглобулінів, однак такий підхід є неінформативним при дефіциті міноритарних субкласів IgG [68]. Золотим стандартом діагностики дефіциту субкласів IgG вважають ELISA [171], хоча деякі автори застосовували радіальну імунодифузію, яка відома ще з 60-х років минулого сторіччя [38]. Наразі вважають, що за допомогою простої радіальної імунодифузії можна коректно діагностувати лише тотальний дефіцит IgG та дефіцит IgG1 [166]. Твердофазний імуноферментний аналіз застосовується як альтернатива ELISA [142]. Деякі автори використовували нефелометрію [94], однак точність результатів цього методу поступається ELISA. B. Bozkurt зі співавт. застосовували зворотний імуноферментний аналіз зі сендвічною технікою ELISA [20]. Радіоімунний метод вважається найточнішим, однак він є недоступним у багатьох клінічних центрах [32]. У разі превалювання пневмококової інфекції рекомендовано додатково вивчати поствакцинальний імунітет після введення полівалентної антипневмококової вакцини (діагностичний рівень < 0,035 мг/мл) [155]. A. Depiero зі співавт. рекомендують одночасне використання кількох лабораторних методів при верифікації діагнозу, зокрема нефелометрії, імуноферментного аналізу та радіальної імунодифузії [35].
Наразі превалює формальна діагностика — за відхиленням виміряної сироваткової концентрації субкласу IgG від нормативних діапазонів. Так, для 7-річної дитини встановлені такі референтні значення: IgG1 = 280–1120 мг/дл, IgG2 = 30–630 мг/дл, IgG3 = 40–250 мг/дл, IgG4 = 11–620 мг/дл [178]. Натомість P.G. Shackelford зі співавт. пропонують діагностувати дефіцит субкласів при зниженні вмісту імуноглобулінів на 3SD від медіани вікової географічної норми [150]. Іноді існує потреба у розрахунку індексу IgG2/IgG для діагностики прихованого дефіциту IgG2. Southern blot може застосовуватися для виявлення делецій у генах важких ланцюгів імуноглобулінів у разі первинного дефіциту субкласів IgG [164].
Диференціальна діагностика
При комбінованому дефіциті субкласів IgG2 та IgG4 слід проводити диференціальну діагностику з синдромами Луї — Барр та Віскотта — Олдрича, особливо в разі поєднання з дефіцитом IgА [112]. При переважанні у клінічній картині пневмококової інфекції здійснюють диференціальну діагностику з дефіцитом специфічних антитіл [118]. C. Brägger зі співавт. описали дефіцит IgG2 і IgG4 в пацієнта з хронічним слизовошкірним кандидозом і гіпотиреоїдизмом. Замісна в/в імуноглобулінотерапія призвела до припинення рецидивних пневмоній у цієї дитини [22]. Слід враховувати, що при дефіциті субкласів IgG, особливо при поєднаних ураженнях, може мати місце прихований дефіцит специфічних антитіл до полісахаридних антигенів. Зокрема, J.H. Braconier зі співавт. виявили дефіцит специфічних IgM до пневмококових капсулярних антигенів у пацієнта з рецидивною пневмококовою інфекцію, у якого відзначався дефіцит IgG2, IgG4 та IgA, однак був нормальний рівень загального IgM [21]. У пацієнтів із тяжким дефіцитом субкласів IgG слід подумати про субкомпенсовану форму загального варіабельного імунодефіциту та хвороби Брутона. Клінічна картина загального варіабельного імунодефіциту й дефіциту субкласів IgG дуже подібна, хоча в першому разі відзначається вірогідно щільніша асоціація з автоімунними ускладненнями, особливо синдромом Шегрена та тиреоїдитом Хашимото, як показали J.C. Barton зі співавт. [11].
Лікування
Як зазначає R.G. Fadal, для пацієнтів з м’якими формами імунодефіциту може бути корисною профілактична антибіотикотерапія, тоді як особи з глибоким зниженням сироваткової концентрації субкласів IgG, що супроводжується тяжкими клінічними симптомами, мають отримувати в/в імуноглобулінотерапію [39].
N.E. Karaca зі співавт. показали користь від профілактичної антибіотикотерапії пролонгованим бензилпеніциліном та антибіотиками короткої дії у дітей із дефіцитом субкласів IgG у ретроспективному дослідженні. Частота інфекційних епізодів знизилася з 12,4 до 5,7 випадку на рік [74]. N. Kutukculer зі співавт. застосовували бензатин пеніциліну, пероральні антибіотики і препарати бактеріальних лізатів для профілактики інфекційних епізодів у пацієнтів із поєднаними формами імунодефіциту (n = 68) і показали однакову ефективність трьох превентивних стратегій [87]. Існують поодинокі клінічні повідомлення про компенсацію антипневмококового гуморального імунітету у дітей із дефіцитом субкласів IgG після імунізації полівалентною пневмококовою вакциною [118].
B.E. Del-Rio-Navarro зі співавт. провели подвійне сліпе плацебо-контрольоване випробування OM-85 BV в дозі 3,5 мг щодня 10 діб поспіль кожного місяця протягом 3 місяців при рецидивних інфекціях верхніх дихальних шляхів, асоційованих із субнормальними рівнями субкласів IgG. Показано вірогідне зменшення частоти інфекційних епізодів у групі терапії [34]. F. Genel, N. Kutukculer провели рандомізоване проспективне дослідження, результати якого показали однакову клінічну ефективність OM-85 BV і профілактичної антибіотикотерапії при дефіциті субкласів IgG, однак 10 із 99 пацієнтів потребували проведення в/в імуноглобулінотерапії, оскільки не відповідали на обидві терапевтичні стратегії [49].
Навіть низькодозова терапія глюкокортикостероїдами призводить до поглиблення імунодефіциту й унеможливлює спонтанну або індуковану компенсацію імунної дисфункції, хоча й послаблює прояви алергічних або автоімунних ускладнень [42].
Імуноглобулінотерапія має бути призначена тяжким пацієнтам із дефіцитом субкласів IgG як засіб лікування першої лінії та як компонент терапії другої лінії після невдачі профілактичної антибіотикотерапії в пацієнтів із легкими й помірними клінічними проявами. Запропоновано застосовувати імуноглобуліни для в/м та в/в введення, причому останні мають більшу доказову базу ефективності.
Повідомлення про клінічні випадки вказують на очевидну користь від застосування в/м нормального імуноглобуліну при дефіциті субкласів IgG [128]. T. Söderström зі співавт. провели проспективне дослідження, присвячене оцінці ефективності препарату імуноглобуліну для в/м введення в дозі 25 мг/кг/тижд протягом 1 року в дорослих пацієнтів із дефіцитом окремих субкласів IgG (n = 43). Показано, що під впливом проведеної імунотерапії досягли вірогідного зменшення кількості днів за рік, під час яких відзначалися інфекційні епізоди. Сироваткові концентрації всіх субкласів IgG були нормалізовані у 24 із 43 пацієнтів. Інші 19 хворих надалі отримували в/м імуноглобулін у дозі 50 мг/кг/тижд, що забезпечило досягнення клініко-лабораторної ремісії [154].
Повідомлення про клінічні випадки вказують на очевидну користь від застосування в/в імуноглобуліну при дефіциті субкласів IgG [115, 144, 151, 165]. Препарат застосовується в дозі 300–400 мг/кг маси тіла кожні 3–4 тижні [112]. Виняток становить лише дефіцит IgG3, коли в/в імуноглобулін слід вводити в дозі 100 мг/кг/тиждень, зважаючи на короткий період напівжиття цього субкласу IgG [121].
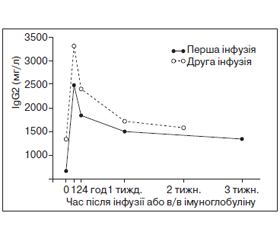
Клінічна ефективність замісної в/в імуноглобулінотерапії продемонстрована в кількох контрольованих та неконтрольованих випробуваннях [1, 2, 14, 18, 54]. В/в імуноглобулін покращує якість життя, знижує кількість інфекційних епізодів та потребу в антибіотиках, а також відновлює сироваткову концентрацію субкласів IgG у пацієнтів із цією імунною дисфункцією [1] (рис. 9).
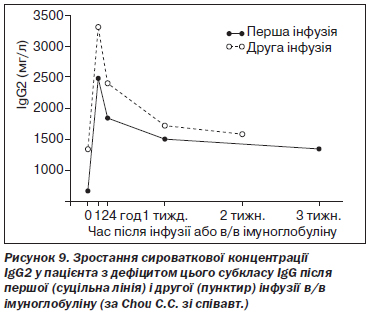
E. Bernatowska-Matuszkiewicz зі співавт. провели проспективне контрольоване випробування дворічної замісної в/в імуноглобулінотерапії в дозі 400 мг/кг/міс у пацієнтів із дефіцитом субкласів IgG, що проявлявся рецидивними респіраторними інфекціями. Показано скорочення тривалості перебування в стаціонарі (перший рік — 27,8 дня, другий рік — 4,9 дня), використання антибіотиків (132,8 проти 30,9 дня) та стероїдів (21,4 проти 0,7 дня) [16]. C.L. Gordon зі співавт. у спеціально спланованому дослідженні продемонстрували ефективність в/в імуноглобуліну для лікування пневмонії, викликаної вірусом грипу H1N1, у пацієнтів з ізольованим дефіцитом IgG2 [52]. A.M. Olinder-Nielsen зі співавт. провели ретроспективний аналіз застосування в/в імуноглобуліну в дозі 100 мг/кг/тиждень у 350 пацієнтів із дефіцитом субкласів IgG, які страждали від рецидивних респіраторних інфекцій. Досягнута редукція кількості інфекційних епізодів, що вимагали призначення антибіотиків, при дефіциті IgG1 — на 57 %, IgG2 — на 59 %, IgG3 — на 63 % та при комбінованих порушеннях — на 61 % (p < 0,001) [121].
Імунотерапія допомагає також при автоімунних і алергічних ускладненнях імунодефіциту. L.F. Bertoli зі співавт. продемонстрували покращення слуху після призначення в/в імуноглобуліну в 60,7 % випадків серед пацієнтів із дефіцитом субкласів IgG1 і IgG3, які страждали від автоімунної сенсоневральної приглухуватості [17]. L.P. Frohman зі співавт. вилікували рецидивний стероїдзалежний оптичний неврит у пацієнта із дефіцитом IgG2 та IgG3 за рахунок в/в імуноглобулінотерапії [45]. S. Chinratanapisit зі співавт. усунули прояви рефрактерного риносинуситу і назального поліпозу в більшості пацієнтів із дефіцитом субкласів IgG під впливом в/в імуноглобуліну [26]. Як зазначають A. May зі співавт., ендоназальна хірургія має застосовуватися тільки при неефективності консервативних підходів до лікування, включаючи в/в імуноглобулін [107]. Згідно з результатами дослідження R. Page зі співавт., відзначається суттєве зменшення клінічних проявів бронхіальної астми під впливом в/в імуноглобуліну у пацієнтів із дефіцитом субкласів IgG [127]. C. Loza Cortina усунув прояви стероїдзалежної астми під впливом високодозової імуноглобулінотерапії у пацієнта з ізольованим дефіцитом IgG2 [101]. Натомість A. Plebani зі співавт. продемонстрували послаблення симптомів рефрактерної епілепсії у дітей із цим імунодефіцитом після призначення в/в імуноглобуліну [135]. Відповідно до цього, M. Sterio зі співавт. повідомили про купірування епілептичного синдрому у 2 дітей із дефіцитом IgG2 після призначення в/в імуноглобуліну в дозі 400 мг/кг/міс [157]. V. Popa зі співавт. продемонстрували усунення проявів хронічної інтерстиціальної хвороби легень, зумовленої дефіцитом субкласів IgG, під впливом в/в імуноглобуліну [136].
S. Suga зі співавт. повідомили про успішну пересадку кісткового мозку 8-річному хлопчику з тяжким дефіцитом IgG1 від HLA-ідентичної MLC-негативної сестри, оскільки ні профілактична антибіотикотерапія, ні в/в імуноглобулін не зменшили частоту інфекційних епізодів. Після трансплантації відзначалися відновлення сироваткової концентрації IgG1 і повна компенсація клінічного статусу [159]. Однак, як показали S.M. Kelsey зі співавт. у спеціально спланованому дослідженні, вторинні дефіцити субкласів IgG можуть персистувати понад 1 рік після пересадки алогенного кісткового мозку у людей і зумовлювати розвиток ускладнень [78]. L. Hammarström, C.I. Smith повідомили про перенесення реципієнту, який раніше не страждав від первинного імунодефіциту, генетично детермінованого дефіциту IgG2 від донора внаслідок проведення трансплантації алогенного кісткового мозку [56].
1. Abdou N.I., Greenwell C.A., Mehta R. et al. Efficacy of intravenous gammaglobulin for immunoglobulin G subclass and/or antibody deficiency in adults // Int. Arch. Allergy Immunol. — 2009. — Vol. 149(3). — P. 267–274.
2. Abrahamian F., Agrawal S., Gupta S. et al. Immunological and clinical profile of adult patients with selective immunoglobulin subclass deficiency: response to intravenous immunoglobulin therapy // Clin. Exp. Immunol. — 2010. — Vol. 159(3). — P. 344–350.
3. Aghamohammadi A., Sedighipour L., Saeed S.E. et al. Alterations in humoral immunity in relatives of patients with common variable immunodeficiency // J. Investig. Allergol. Clin. Immunol. — 2008. — Vol. 18(4). — P. 266–271.
4. Alexander A., Anicito I., Buxbaum J. Gamma heavy chain disease in man. Genomic sequence reveals two noncontiguous deletions in a single gene // J. Clin. Invest. — 1988. — Vol. 82(4). — P. 1244–1252.
5. Ambrozic J., Logar D., Stajer D. et al. Recurrent sepsis and seronegative arthritis in a patient with a selective IgG3 deficiency // Wien. Klin. Wochenschr. — 2000. — Vol. 112(15–16). — P. 735–737.
6. Andres E., Limbach F.X., Kurtz J.E. et al. Primary humoral immunodeficiency (late-onset common variable immunodeficiency) with antinuclear antibodies and selective immunoglobulin deficiency // Am. J. Med. — 2001. — Vol. 111(6). — P. 489–491.
7. Armenaka M., Grizzanti J., Rosenstreich D.L. Serum immunoglobulins and IgG subclass levels in adults with chronic sinusitis: evidence for decreased IgG3 levels // Ann. Allergy. — 1994. — Vol. 72(6) — P. 507–514.
8. Barton J.C., Barton J.C., Bertoli L.F. et al. Predictors of shingles reports at diagnosis of common variable immunodeficiency and selective immunoglobulin G subclass deficiency in 212 Alabama adults // Infect. Dis. Rep. — 2012. — Vol. 4(2). — e34.
9. Barton J.C., Bertoli L.F., Acton R.T. Common variable immunodeficiency and IgG subclass deficiency in central Alabama hemochromatosis probands homozygous for HFE C282Y // Blood. Cells. Mol. Dis. — 2003. — Vol. 31(1). — P. 102–111.
10. Barton J.C., Bertoli L.F., Acton R.T. HLA-A and -B alleles and haplotypes in 240 index patients with common variable immunodeficiency and selective IgG subclass deficiency in central Alabama // BMC. Med. Genet. — 2003. — Vol. 4. — P. 3.
11. Barton J.C., Bertoli L.F., Barton J.C. et al. Comparisons of CVID and IgGSD: referring physicians, autoimmune conditions, pneumovax reactivity, immunoglobulin levels, blood lymphocyte subsets, and HLA-A and -B typing in 432 adult index patients // J. Immunol. Res. — 2014. — 542–706.
12. Barton J.C., Barton J.C., Bertoli L.F. et al. Predictors of shingles reports at diagnosis of common variable immunodeficiency and selective immunoglobulin G subclass deficiency in 212 Alabama adults // Infect. Dis. Rep. — 2012. — Vol. 4(2). — e34.
13. Bass J.L., Nuss R., Mehta K.A. et al. Recurrent meningococcemia associated with IgG2-subclass deficiency // N. Engl. J. Med. — 1983. — Vol. 309(7). — P. 430.
14. Beard L.J., Ferrante A. IgG replacement therapy in IgG subclass-deficient children // Monogr. Allergy. — 1988. — Vol. 23. — P. 194–203.
15. Beck C.S., Heiner D.C. Selective immunoglobulin G4 deficiency and recurrent infections of the respiratory tract // Am. Rev. Respir. Dis. — 1981. — Vol. 124(1). — P. 94–96.
16. Bernatowska-Matuszkiewicz E., Pac M., Skopcynska H. et al. Clinical efficacy of intravenous immunoglobulin in patients with severe inflammatory chest disease and IgG3 subclass deficiency // Clin. Exp. Immunol. — 1991. — Vol. 85(2). — P. 193–197.
17. Bertoli L.F., Pappas D.G., Barton J.C., Barton J.C. Serum immunoglobulins in 28 adults with autoimmune sensorineural hearing loss: increased prevalence of subnormal immunoglobulin G1 and immunoglobulin G3 // BMC. Immunol. — 2014. — Vol. 15. — P. 43.
18. Björkander J., Oxelius V.A., Söderström R., Hanson L.A. Immunoglobulin treatment of patients with selective IgG subclass and IgA deficiency states // Monogr. Allergy. — 1988. — Vol. 23. — P. 160–167.
19. Bos-Veneman N.G., Olieman R., Tobiasova Z. et al. Altered immunoglobulin profiles in children with Tourette syndrome // Brain. Behav. Immun. — 2011. — Vol. 25(3). — P. 532–538.
20. Bozkurt B., Artac H., Arslan N. et al. Systemic atopy and immunoglobulin deficiency in Turkish patients with vernal keratoconjunctivitis // Ocul. Immunol. Inflamm. — 2013. — Vol. 21(1). — P. 28–33.
21. Braconier J.H., Nilsson B., Oxelius V.A., Karup-Pedersen F. Recurrent pneumococcal infections in a patient with lack of specific IgG and IgM pneumococcal antibodies and deficiency of serum IgA, IgG2 and IgG4 // Scand. J. Infect. Dis. — 1984. — Vol. 16(4) — Vol. 407–410.
22. Brägger C., Seger R.A., Aeppli R. et al. IgG2/IgG4 subclass deficiency in a patient with chronic mucocutaneous candidiasis and bronchiectases // Eur. J. Pediatr. — 1989. — Vol. 149(3). — P. 168–169.
23. Caksen H., Oner A.F., Arslan S. et al. Immunoglobulin subgroups in children with febrile seizures // Pediatr. Int. — 2001. — Vol. 43(1). — P. 58–60.
24. Carvalho Neves Forte W., Ferreira De Carvalho Junior F., Damaceno N. et al. Evolution of IgA deficiency to IgG subclass deficiency and common variable immunodeficiency // Allergol. Immunopathol. (Madr.). — 2000. — Vol. 28(1). — P. 18–20.
25. Castro R., Robinson N., Klein J., Geimeier W. Malignant external otitis and mastoiditis associated with an IgG4 subclass deficiency in a child // Del. Med. J. — 1990. — Vol. 62(12). — P. 1417–1421.
26. Chinratanapisit S., Tunsuriyawong P., Vichyanond P. et al. Chronic rhinosinusitis and recurrent nasal polyps in two children with IgG subclass deficiency and review of the literature // J. Med. Assoc. Thai. — 2005. — Vol. 88(8). — S251–258.
27. Collins A.M., Jackson K.J.L. A temporal model of human IgE and IgG antibody function // Frontiers in Immuno–logy. — 2013. — Vol. 4. — P. 235.
28. Cunningham-Rundles C., Fotino M., Rosina O., Peter J.B. Selective IgA deficiency, IgG subclass deficiency, and the major histocompatibility complex // Clin. Immunol. Immunopathol. — 1991. — Vol. 61(2 Pt 2). — S61–69.
29. Dahlen G., Björkander J., Gahnberg L. et al. Periodontal disease and dental caries in relation to primary IgG subclass and other humoral immunodeficiencies // J. Clin. Periodontol. — 1993. — Vol. 20(1). — P. 7–13.
30. De Gracia J., Rodrigo M.J., Morell F. et al. IgG subclass deficiencies associated with bronchiectasis // Am. J. Respir. Crit. Care Med. — 1996. — Vol. 153(2). — P. 650–655.
31. De José Gomez M.I., González de Dios J., Hernando de Larramendi C. et al. IgG2 deficiency associated with recurrent pneumonia and asthma (review of an IgG subclass) // An. Esp. Pediatr. — 1990. — P. 33(3). — P. 258–264.
32. De Moraes Lui C., Oliveira L.C., Diogo C.L. et al. Immunoglobulin G subclass concentrations and infections in children and adolescents with severe asthma // Pediatr. Allergy Immunol. — 2002. — Vol. 13(3). — P. 195–202.
33. DeBaets F., Kint J., Pauwels R., Leroy J. IgG subclass deficiency in children with recurrent bronchitis // Eur. J. Pediatr. — 1992. — Vol. 151(4) — P. 274–278.
34. Del-Rio-Navarro B.E., Luis Sienra-Monge J.J., Berber A. et al. Use of OM-85 BV in children suffering from recurrent respiratory tract infections and subnormal IgG subclass levels // Allergol. Immunopathol. (Madr.). — 2003. — Vol. 31(1) — P. 7–13.
35. Depiero A., Kaminski D.A., Halsey J.F. et al. Immunologic compensation in a patient with a large IgH constant region deletion // J Allergy Clin. Immunol. — 2001. — Vol. 107(6). — P. 1051–1055.
36. Düzgün N., Peksari Y., Sonel B. et al. Localization of extrapulmonary tuberculosis in the synovial membrane, skin, and meninges in a patient with systemic lupus erythematosus and IgG deficiency // Rheumatol. Int. — 2002. — Vol. 22(1). — P. 41–44.
37. Eriksson P., Almroth G, Denneberg T., Lindström F.D. IgG2 deficiency in primary Sjogren’s syndrome and hypergammaglobulinemic purpura // Clin. Immunol. Immunopathol. — 1994. — Vol. 70(1). — P. 60–65.
38. Escobar-Pérez X., Dorta-Contreras A.J., Interián-Morales M.T. et al. IgG2 immunodeficiency: association to pediatric patients with bacterial meningoencephalitis // Arq. Neuropsiquiatr. — 2000. — Vol. 58(1). — P. 141–145.
39. Fadal R.G. Chronic sinusitis, steroid-dependent asthma, and IgG subclass and selective antibody deficiencies // Otolaryngol. Head. Neck. Surg. — 1993. — Vol. 109(3 Pt 2). — P. 606–610.
40. Faller J.P., Ruyer O., Kara A. et al. Discovery of IgG subclass deficiency in a case of meningococcal purpura fulminans // Presse Med. — 1992. — Vol. 21(5). — P. 220.
41. Farmer J.R., Sokol C.L., Bonilla F.A. et al. Bilateral lung transplantation in a patient with humoral immune deficiency: a case report with review of the literature // Case Reports Immunol. — 2014. — 910215.
42. Fedor M.E., Rubinstein A. Effects of long-term low-dose corticosteroid therapy on humoral immunity // Ann. Allergy Asthma Immunol. — 2006. — Vol. 97(1). — P. 113–116.
43. Feldman C., Weltman M., Wadee A. et al. A study of immunoglobulin G subclass levels in black and white patients with various forms of obstructive lung disease // S. Afr. Med. J. — 1993. — Vol. 83(1). — P. 9–12.
44. Freeman J.A., Crassini K.R., Best O.G. et al. Immunoglobulin G subclass deficiency and infection risk in 150 patients with chronic lymphocytic leukemia // Leuk. Lymphoma. — 2013. — Vol. 54(1). — P. 99–104.
45. Frohman L.P., Cook S.D., Bielory L. et al. Dysgammaglobulinemia in steroid-dependent optic neuritis: response to gammaglobulin treatment // J. Clin. Neuroophthalmol. — 1991. — Vol. 11(4). — P. 241–245.
46. Gallina R., Bottaro A., Boccazzi C. et al. The genetics of IgG4 deficiency: role of the immunoglobulin heavy chain constant region and HLA loci // Eur. J. Immunol. — 1992. — Vol. 22(1). — P. 227–233.
47. Garg V., Lipka S., Rizvon K. et al. Diffuse nodular lymphoid hyperplasia of intestine in selective IgG 2 subclass deficiency, autoimmune thyroiditis, and autoimmune hemolytic anemia: case report and literature review // J. Gastrointestin. Liver Dis. — 2012. –Vol. 21(4). — P. 431–434.
48. Garty B.Z. Enteropathy and IgG subclass deficiency // J. Pediatr. — 1996. –– Vol. 128(5 Pt 1). — P. 722–723.
49. Genel F., Kutukculer N. Prospective, randomized comparison of OM-85 BV and a prophylactic antibiotic in children with recurrent infections and immunoglobulin A and/or G subclass deficiency // Curr. Ther. Res. Clin. Exp. — 2003. — Vol. 64(8). — P. 600–615.
50. Go T. Carbamazepine-induced IgG1 and IgG2 deficiency associated with B cell maturation defect // Seizure. — 2004. — Vol. 13(3). — P. 187–190.
51. Gordon C.L., Johnson P.D., Permezel M. et al. Association between severe pandemic 2009 influenza A (H1N1) virus infection and immunoglobulin G(2) subclass deficiency // Clin. Infect. Dis. — 2010. — Vol. 50(5). — P. 672–678.
52. Gordon C.L., Langan K., Charles P.G. et al. Pooled human immunoglobulin therapy in critically Ill patients with pandemic 2009 influenza A(H1N1) pneumonitis and immunoglobulin G2 subclass (IgG2) deficiency // Clin. Infect. Dis. — 2011. — Vol. 52(3). — P. 422–426.
53. Gottsegen D.N. Pneumococcal osteomyelitis associated with IgG2 subclass deficiency // Pediatr. Infect. Dis. J. — 1987. — Vol. 6(3). — P. 281–284.
54. Grob M., Joller-Jemelka H.I., Grob P.J. et al. Deficiency in immunoglobulin subclasses as cause of increased infection susceptibility — a family study // Schweiz. Med. Wochenschr. — 1991. — Vol. 121(5). — P. 133–144.
55. Hahn-Zoric M., Ulanova M., Friman V. et al. Antibody response to the Haemophilus influenzae type b-tetanus toxoid conjugate vaccine in healthy and infection-prone individuals with IgG3 subclass deficiency // J. Clin. Immunol. — 2004. — Vol. 24(5). — P. 561–570.
56. Hammarström L., Smith C.I. Development of IgG2 deficiency in a bone-marrow-transplanted patient. Implication for generation of the anticarbohydrate antibody repertoire in subclass-deficient individuals // Transplantation. — 1987. — Vol. 43(6). — P. 917–919.
57. Hanson L.A., Söderström R., Nilssen D.E. et al. IgG subclass deficiency with or without IgA deficiency // Clin. Immunol. Immunopathol. — 1991. — Vol. 61(2 Pt 2). — S. 70–77.
58. Hashira S., Okitsu–Negishi S., Yoshino K. Placental transfer of IgG subclasses in a Japanese population // Pediatr. Int. — 2000. — Vol. 42 (4). — P. 337–342.
59. Hassani R.T., Rousseau A., de Monchy I. et al. Retinal vasculitis revealing immunoglobulin G subclass deficiency // Ocul. Immunol. Inflamm. — 2013. — Vol. 21(1). — P. 84–86.
60. Hassett J., Meyers S., McFarland L., Mulligan M.E. Recurrent Clostridium difficile infection in a patient with selective IgG1 deficiency treated with intravenous immune globulin and Saccharomyces boulardii // Clin. Infect. Dis. — 1995. — Vol. 20(2). — S. 266–268.
61. Hoffman J.D., Ciprero K.L., Sullivan K.E. et al. Immune abnormalities are a frequent manifestation of Kabuki syndrome // Am. J. Med. Genet. A. — 2005. — Vol. 135(3). — P. 278–281.
62. Ideura G., Agematsu K., Komatsu Y. et al. Selective IgM deficiency accompanied with IgG4 deficiency, dermal complications and a bronchial polyp // Allergol. Int. — 2008. — Vol. 57(1). — P. 99–105.
63. Inoue R., Kondo N., Kobayashi Y. et al. IgG2 deficiency associated with defects in production of interferon-gamma; comparison with common variable immunodeficiency // Scand. J. Immunol. — 1995. — Vol. 41(2). — P. 130–134.
64. Insel R.A., Anderson P.W. Response to oligosaccharide-protein conjugate vaccine against Hemophilus influenzae b in two patients with IgG2 deficiency unresponsive to capsular polysaccharide vaccine // N. Engl. J. Med. — 1986. — Vol. 315(8). — P. 499–503.
65. Ishizaka A., Nakanishi M., Kasahara E. et al. Phenytoin-induced IgG2 and IgG4 deficiencies in a patient with epilepsy // Acta Paediatr. — 1992. — Vol. 81(8). — P. 646–648.
66. Jiménez A., Alvarez-Doforno R., García Rodríguez M.C. et al. Autoantibodies in patients with IgA and IgG2 deficiencies // APMIS. — 1991. — Vol. 99(4) — P. 327–332.
67. Johnston S.L., Virgo P.F., Unsworth D.J. et al. Type 1 diabetes mellitus masking primary antibody deficiency // J. Clin. Pathol. — 2000. — Vol. 53(3). — P. 236–237.
68. Jolles S., Borrell R., Zouwail S. et al. Calculated globulin (CG) as a screening test for antibody deficiency // Clin. Exp. Immunol. — 2014. — Vol. 177(3). — P. 671–678.
69. Jolles S., Tyrer M., Johnson M., Webster D. et al. Long term recovery of IgG and IgM production during HIV infection in a patient with common variable immunodeficiency (CVID) // J. Clin. Pathol. — 2001. –Vol. 54(9). — P. 713–715.
70. Kallio-Laine K., Seppänen M., Aittoniemi J. et al. HLA-DRB1*01 allele and low plasma immunoglobulin G1 concentration may predispose to herpes-associated recurrent lymphocytic meningitis // Hum. Immunol. — 2010. — Vol. 71(2). — P. 179–181.
71. Kallio-Laine K., Seppänen M., Lokki M.L. et al. Widespread unilateral pain associated with herpes simplex virus infections // J. Pain. — 2008. — Vol. 9(7). — P. 658–665.
72. Kamei K., Nakagawa A., Otsuka Y. et al. Chronic glomerulonephritis associated with IgG subclass deficiency // Pediatr. Nephrol. — 2007. — Vol. 22(8). — P. 1229–1234.
73. Kaneko H., Nakashima M., Kudo A. et al. Selective IgG deficiency with a transcriptional disorder of the gamma switching region gene and the IL–4 gene // Int. Immunol. — 1990. — Vol. 2(7). — P. 661–668.
74. Karaca N.E., Karadeniz C., Aksu G., Kutukculer N. Clinical and laboratory evaluation of periodically monitored Turkish children with IgG subclass deficiencies // Asian. Pac. J. Allergy Immunol. — 2009. — Vol. 27(1). — P. 43–48.
75. Karaman O., Uğuz A., Uzuner N. Immunoglobulin G subclasses in wheezing infants // Acta Paediatr. Jpn. — 1998. Vol. 40(6). — P. 564–566.
76. Kavanaugh A.F., Huston D.P. Variable expression of IgG2 deficiency // J. Allergy. Clin. Immunol. — 1990. — Vol. 86(1). — P. 4–10.
77. Kawamata N., Sakajiri S., Sugimoto K.J. et al. A novel chromosomal translocation t(1;14)(q25;q32) in pre-B acute lymphoblastic leukemia involves the LIM homeodomain protein gene, Lhx4 // Oncogene. — 2002. — Vol. 21(32). — P. 4983–4991.
78. Kelsey S.M., Lowdell M.W., Newland A.C. IgG subclass levels and immune reconstitution after T cell-depleted allogeneic bone marrow transplantation // Clin. Exp. Immunol. — 1990. — Vol. 80(3). — P. 409–412.
79. Kemper M.J., Meyer-Jark T., Müller-Wiefel D.E. IgG2 deficiency in uremic children is not restricted to peritoneal dialysis treatment // Pediatr. Nephrol. — 1997. — Vol. 11(6). — P. 684–616.
80. Kim J.H., Park H.J., Choi G.S. et al. Immunoglobulin G subclass deficiency is the major phenotype of primary immunodeficiency in a Korean adult cohort // J. Korean. Med. Sci. — 2010. — Vol. 25(6). — P. 824–828.
81. Kirsch I.R., Morton C.C., Nakahara K., Leder P. Human immunoglobulin heavy chain genes map to a region of translocations in malignant B lymphocytes // Science. — 1982. — Vol. 216(4543). — P. 301–303.
82. Klingebiel T., Pickert A., Dopfer R. et al. Unusual course of a Chlamydia pneumonia in an infant with IgG2/IgG4–deficiency // Eur. J. Pediatr. — 1989. — Vol. 148(5). — P. 431–434.
83. Komaroff A.L., Geiger A.M., Wormsely S. IgG subclass deficiencies in chronic fatigue syndrome // Lancet. — 1988. — Vol. 1(8597). — P. 1288–1289.
84. Kondo N., Inoue R., Kasahara K. et al. Failure of IgG production due to a defect in the opening of the chromatin structure of I gamma 1 region in a patient with IgG and IgA deficiency // Clin. Exp. Immunol. — 1995. — Vol. 99(1). — P. 21–28.
85. Kubota M., Yasunaga M., Hashimoto H. et al. IgG4 deficiency with Rothmund-Thomson syndrome: a case report // Eur. J. Pediatr. — 1993. — Vol. 152(5). — P. 406–408.
86. Kuijpers T.W., Weening R.S., Out T.A. IgG subclass deficiencies and recurrent pyogenic infections, unresponsiveness against bacterial polysaccharide antigens // Allergol. Immunopathol. (Madr.). — 1992. — Vol. 20(1). — P. 28–34.
87. Kutukculer N., Karaca N.E., Demircioglu O., Aksu G. et al. Increases in serum immunoglobulins to age-related normal levels in children with IgA and/or IgG subclass deficiency // Pediatr. Allergy Immunol. — 2007. — Vol. 18(2). — P. 167–173.
88. Lacombe C., Aucouturier P., Preud’homme J.L. Selective IgG1 deficiency // Clin. Immunol. Immunopathol. — 1997. — Vol. 84(2). — P. 194–201.
89. Layward L., Allen A.C., Harper S.J. et al. Deficiency of IgG subclass antibody response to tetanus toxoid associated with high serum IgA levels in IgA nephropathy // Clin. Nephrol. — 1993. — Vol. 40(3) — P. 131–136.
90. Lefranc G., Chaabani H., Van Loghem E. et al. Simultaneous absence of the human IgG1, IgG2, IgG4 and IgA1 subclasses: immunological and immunogenetical considerations // Eur. J. Immunol. — 1983. — Vol. 13(3). — P. 240–244.
91. Leickly F.E., Buckley R.H. Development of IgA and IgG2 subclass deficiency after sulfasalazine therapy // J. Pediatr. — 1986. — Vol. 108(3). — P. 481–482.
92. Lenoir G.M., Preud’homme J.L., Bernheim A., Berger R. Correlation between immunoglobulin light chain expression and variant translocation in Burkitt’s lymphoma // Nature. — 1982. — Vol. 298(5873). — P. 474–476.
93. Lenti C., Masserini C., Barlocco A. et al. IgG2 deficiency in children with febrile convulsions: a familial study // Ital. J. Neurol. Sci. — 1993. — Vol. 14(7). — P. 561–564.
94. Levendoglu F., Reisli I., Ustun M.E. et al. Spinal epidural abscess associated with IgG4 deficiency // J. Spinal. Disord. Tech. — 2003. — Vol. 16(1). — P. 104–107.
95. Likosky D.J., Kraus E.E., Yuen E.C. Recurrent multifocal demyelinating neuropathy with febrile illness and IgG subset deficiency // Neurology. — 1999. — Vol. 52(9). — P. 1902–1905.
96. Linde A., Söderström R., Smith C.I. et al. Herpesvirus serology, aberrant specific immunoglobulin G2 and G3 subclass patterns and Gm allotypes in individuals with low levels of IgG3 // Clin. Exp. Immunol. — 1992. — Vol. 90(2). — P. 199–203.
97. Litzman J., Bartonkovą D., Pikulovą Z. et al. IgG subclasses and autoantibodies in adult patients with selective IgA deficiency // Vnitr. Lek. — 2000. — Vol. 46(3). — P. 170–173.
98. Lock K., Anders S., Ernst M. et al. Immunoglobulin G subclass deficiency in patients with asthma and chronic obstructive bronchitis // Immun. Infekt. — 1990. — Vol. 18(5). — P. 157–161.
99. Loftus B.G., Price J.F., Lobo-Yeo A., Vergani D. IgG subclass deficiency in asthma // Arch. Dis. Child. — 1988. — Vol. 63(12). — P. 1434–1437.
100. Loh R.K., Thong Y.H., Ferrante A. Immunoglobulin G subclass deficiency in children with high levels of immunoglobulin E and infection proneness // Int. Arch. Allergy Appl. Immunol. — 1990. — Vol. 93(4). — P. 285–258.
101. Loza Cortina C. Severe steroid-dependent asthma with IgG-2 deficiency and recurrent sinusitis: response to treatment with high-dose intravenous immunoglobulin // Allergol. Immunopathol. (Madr.). — 1999. — Vol. 27(3). — P. 165–167.
102. Maak B., Müller E., Berbig H., Estel C. Maternal deficiency of IgG 2 and IgG 4 and neonatal infection caused by B–streptococci // Zentralbl. Gynakol. — 1993. — Vol. 115(3). — P. 136–139.
103. Maeoka Y., Hara T., Dejima S., Takeshita K. IgA and IgG2 deficiency associated with zonisamide therapy: a case report // Epilepsia. — 1997. – Vol. 38(5). — P. 611–613.
104. Maes M., Coucke F., Leunis J.C. Normalization of the increased translocation of endotoxin from gram negative enterobacteria (leaky gut) is accompanied by a remission of chronic fatigue syndrome // Neuro Endocrinol Lett. — 2007. — Vol. 28(6). — P. 739–744.
105. Martinot M., Oswald L., Parisi E. et al. Immunoglobulin deficiency in patients with Streptococcus pneumoniae or Haemophilus influenzae invasive infections // Int. J. Infect. Dis. — 2014. — Vol. 19. — P. 79–84.
106. Matter L., Wilhelm J.A., Angehrn W. et al. Selective antibody deficiency and recurrent pneumococcal bacteremia in a patient with Sjögren’s syndrome, hyperimmunoglobulinemia G, and deficiencies of IgG2 and IgG4 // N. Engl. J. Med. — 1985. — Vol. 312(16). — P. 1039–1042.
107. May A., Zielen S., Reimold I. et al. Immunoglobulin subclass defects in patients with therapy refractor chronic rhinosinusitis // HNO. — 1999. — Vol. 47(1). — P. 19–24.
108. Meyts I., Jansen K., Renard M. et al. Neuromyelitis optica-IgG+ optic neuritis associated with celiac disease and dysgammaglobulinemia: a role for tacrolimus? // Eur. J. Paediatr. Neurol. — 2011. — Vol. 15(3). — P. 265–267.
109. Mikhak Z., Mulliken J.B., Lee J. et al. Humoral immune deficiency and hemifacial microsomia seen in one family // Cleft. Palate. Craniofac. J. — 2009. — Vol. 46(5). — P. 477–480.
110. Mitsui H., Komine M., Shirai A. et al. Chronic active EB virus infection complicated with IgG3 subclass deficiency: an adult case treated with intravenous immunoglobulin and IFN-alpha // Acta Derm. Venereol. 2003. — 83(1). — P. 31–35.
111. Miwa Y., Inoue R., Ozawa T. et al. IgG subclass levels and southern analysis of DNA in primary immunodeficiency diseases including IgG subclass deficiency // Exp. Clin. Immunogenet. — 1994. — Vol. 11(4). — P. 173–181.
112. Morell A. Clinical relevance of IgG subclass deficiencies // Ann. Biol. Clin. (Paris). — 1994. — Vol. 52(1). — P. 49–52.
113. Moss R.B., Carmack M.A., Esrig S. Deficiency of IgG4 in children: association of isolated IgG4 deficiency with recurrent respiratory tract infection // J. Pediatr. — 1992. — Vol. 120(1) — P. 16–21.
114. Natta C.L., Outschoorn I.M. IgG2 deficiency in sickle cell anaemia // Scand. J. Haematol. — 1984. — Vol. 33(2). — P. 129–134.
115. Nettagul R., Visitsunthorn N., Vichyanond P. et al. A case of IgG subclass deficiency with the initial presentation of transient hypogammaimmunoglobulinemia of infancy and a review of IgG subclass deficiencies // J. Med. Assoc. Thai. — 2003. — Vol. 86(7). — P. 686–692.
116. Nieto M., Roldán S., Sánchez B. et al. Immunological study in patients with severe myoclonic epilepsy in childhood // Rev. Neurol. — 2000. — Vol. 30(5). — P. 412–414.
117. Ninomiya H., Hasegawa H., Fukuoka T. et al. Selective IgG2,4 subclass and IgE deficiencies in an adult patient with recurrent pneumonia // Jpn. J. Med. — 1991. — Vol. 30(4). — P. 338–342.
118. Ohga S., Okada K., Asahi T. et al. Recurrent pneumococcal meningitis in a patient with transient IgG subclass deficiency // Acta Paediatr. Jpn. — 1995. — Vol. 37(2). — P. 196–200.
119. Ojuawo A., Milla P.J., Lindley K.J. Serum immunoglobulin and immunoglobulin G subclasses in children with allergic colitis // West. Afr. J. Med. — 1998. Vol. 17(3) — P. 206–209.
120. O’Keeffe S., Gzel A., Drury R. et al. Immunoglobulin G subclasses and spirometry in patients with chronic obstructive pulmonary disease // Eur. Respir. J. — 1991. — Vol. 4(8). — P. 932–936.
121. Olinder-Nielsen A.M., Granert C., Forsberg P. et al. Immunoglobulin prophylaxis in 350 adults with IgG subclass deficiency and recurrent respiratory tract infections: a long-term follow-up // Scand. J. Infect. Dis. — 2007. — Vol. 39(1). — P. 44–50.
122. Outschoorn I.M., Natta C.L. IgG subclass alterations in adult asthma // Microbiol. Immunol. — 1992. — Vol. 36(9) — P. 977–982.
123. Ovesen T., Kragelund J.R., Jensen J.M. et al. Immunodeficiencies in children with chronic post tympanic –otorrhoea // Dan. Med. Bull. — 2011. — Vol. 58(7). — A4282.
124. Oxelius V.A., Hanson L.A., Björkander J. et al. IgG3 deficiency: common in obstructive lung disease. Hereditary in families with immunodeficiency and autoimmune disease // Monogr. Allergy. — 1986. — Vol. 20. — P. 106–115.
125. Oxelius V.A., Lindán V., Christensen K.K., Christensen P. Deficiency of IgG subclasses in mothers of infants with group B streptococcal septicemia // Int. Arch. Allergy. Appl. Immunol. — 1983. — Vol. 72(3). — P. 249–252.
126. Oymak O. IgM nephropathy in a patient with combined deficiency of IgA, IgG2 and IgG4 // Clin. Nephrol. — 1997. — Vol. 47(3). — P. 202–203.
127. Page R., Friday G., Stillwagon P. et al. Asthma and selective immunoglobulin subclass deficiency: improvement of asthma after immunoglobulin replacement therapy // J. Pediatr. — 1988. — Vol. 112(1). — P. 127–131.
128. Pardillos Tomé A., Marcilla Córdoba F., Boldova Gil I., Pac Sa J. Classic Kaposi sarcoma associated with IgG deficiency // Med. Clin. (Barc.). — 2013. — Vol. 140(2). — e3.
129. Pauly E., Linderkamp O., Pöschl J. Incontinentia pigmenti in combination with decreased IgG subclass concentrations in a female newborn // Biol. Neonate. — 2005. — Vol. 88(3). — P. 172–174.
130. Pavone P., Falsaperla R., De Silva K. et al. Down syndrome and arterial ischemic stroke in childhood: a potential immunologic link with selective IgG4 subclass deficiency // Eur. J. Paediatr. Neurol. — 2014. — Vol. 18(4). — P. 520–525.
131. Pengsaa K., Sirivichayakul C., Vithayasai N. et al. Chronic diarrhea and abnormal serum immunoglobulin levels: a case report // Southeast. Asian. J. Trop. Med. Public. Health. — 2007. — Vol. 38(3). — P. 424–426.
132. Péron S., Metin A., Gardès P. et al. Human PMS2 deficiency is associated with impaired immunoglobulin class switch recombination // J. Exp. Med. — 2008. — Vol. 205(11). — P. 2465–2472.
133. Plebani A., Duse M., Tiberti S. Intravenous gamma-globulin therapy and serum IgG subclass levels in intractable childhood epilepsy // Monogr. Allergy. — 1988. — Vol. 23. — P. 204–215.
134. Plebani A., Ugazio A.G., Meini A. et al. Extensive deletion of immunoglobulin heavy chain constant region genes in the absence of recurrent infections: when is IgG subclass deficiency clinically relevant? // Clin. Immunol. Immunopathol. — 1993. — Vol. 68(1). — P. 46–50.
135. Popa V. Airway obstruction in adults with recurrent respiratory infections and IgG deficiency // Chest. — 1994. — Vol. 105(4). — P. 1066–1072.
136. Popa V., Colby T.V., Reich S.B. Pulmonary interstitial disease in Ig deficiency // Chest. — 2002. — Vol. 122(5). — P. 1594–1603.
137. Popa V., Kim K., Heiner D.C. IgG deficiency in adults with recurrent respiratory infections // Ann. Allergy. — 1993. — Vol. 70(5). — P. 418–424.
138. Rahiminejad M.S., Mirmohammad Sadeghi M., Mohammadinejad P. et al. Evaluation of humoral immune function in patients with chronic idiopathic thrombocytopenic purpura // Iran. J. Allergy Asthma Immunol. — 2013. — Vol. 12(1). — P. 50–56.
139. Rawat A., Suri D., Gupta A. et al. Isolated immunoglobulin G4 subclass deficiency in a child with bronchiectasis // Indian. J. Pediatr. — 2014. — Vol. 81(9). — P. 932–933.
140. Read R., Spickett G., Harvey J. et al. IgG1 subclass deficiency in patients with chronic fatigue syndrome // Lancet. — 1988. — Vol. 1(8579). — P. 241–242.
141. Resman F., Svensjö T., Ünal C. et al. Necrotizing myositis and septic shock caused by Haemophilus influenzae type f in a previously healthy man diagnosed with an IgG3 and a mannose-binding lectin deficiency // Scand. J. Infect. Dis. — 2011. — Vol. 43(11–12). — P. 972–976.
142. Roberton D.M., Colgan T., Ferrante A. et al. IgG subclass concentrations in absolute, partial and transient IgA deficiency in childhood // Pediatr. Infect. Dis J. — 1990. — Vol. 9(8). — S. 41–45.
143. Röther E., Vaith P., Peter H.H. Isolated immunoglobulin G (IgG) deficiency in dystrophia myotonica (Curschmann-Steinert) // Med. Klin. (Munich). — 1990. — Vol. 85(1). — 150–153.
144. Saleem T., Rabbani M., Jamil B. et al. Primary IgA and IgG subclass deficiency in a 17-year-old Pakistani girl: a case report // Cases J. — 2009. — Vol. 10(2). — P. 67–85.
145. Santaella M.L., Peredo R., Disdier O.M. et al. IgA deficiency: clinical correlates with IgG subclass and mannan-binding lectin deficiencies // P. R. Health Sci. J. — 2005. — Vol. 24(2). — P. 107–110.
146. Schröder C.H., Bakkeren J.A., Weemaes C.M., Monnens L.A. IgG2 deficiency in young children treated with continuous ambulatory peritoneal dialysis (CAPD) // Perit. Dial. Int. — 1989. — Vol. 9(4). — P. 261–265.
147. Scoular A., Corcoran G.D., Malin A. et al. Fusobacterium nucleatum bacteraemia with multiple liver abscesses in an HIV-I antibody positive man with IgG2 deficiency //J. Infect. — 1992. — Vol. 24(3). — P. 321–325.
148. Seidel M.G., Duerr C., Woutsas S. et al. A novel immunodeficiency syndrome associated with partial trisomy 19p13 // J. Med. Genet. — 2014. — Vol. 51(4) — P. 254–263.
149. Sekerel B.E., Saraçlar Y., Sanal O. et al. IgG subclasses in children with recurrent respiratory tract infections in an allergy practice // Acta Paediatr. Jpn. — 1996. — Vol. 38(2). — P. 124–127.
150. Shackelford P.G., Polmar S.H., Mayus J.L. et al. Spectrum of IgG2 subclass deficiency in children with recurrent infections: prospective study // J. Pediatr. — 1986. — Vol. 108(5Pt1). — P. 647–653.
151. Shield J.P., Strobel S., Levinsky R.J., Morgan G. et al. Immunodeficiency presenting as hypergammaglobulinaemia with IgG2 subclass deficiency // Lancet. — 1992. — Vol. 340(8817). — P. 448–450.
152. Snowden J.A., Milford-Ward A., Cookson L.J., McKendrick M.W. Recurrent lymphocytic meningitis associated with hereditary isolated IgG subclass 3 deficiency // J. Infect. — 1993. — Vol. 27(3). — P. 285–289.
153. Snowden J.A., Milford-Ward A., Reilly J.T. Symptomatic IgG3 deficiency successfully treated with intravenous immunoglobulin therapy // Postgrad. Med. J. — 1994. — Vol. 70(830). — P. 924–926.
154. Söderström T., Söderström R., Enskog A. Immunoglobulin subclasses and prophylactic use of immunoglobulin in immunoglobulin G subclass deficiency // Cancer. — 1991. — Vol. 68(6). — P. 1426–1429.
155. Stanford E., Ladhani S., Borrow R. et al. Immunoglobulin G deficiency in United kingdom children with invasive pneumococcal disease // Pediatr. Infect. Dis. J. — 2011. — Vol. 30(6). — P. 462–465.
156. Stead A., Douglas J.G., Broadfoot C.J. et al. Humoral immunity and bronchiectasis // Clin. Exp. Immunol. — 2002. — Vol. 130(2). — P. 325–330.
157. Sterio M., Gebauer E., Vucićević G. et al. 3 case reports of patients with malignant epilepsy treated with high doses of intravenous immunoglobulin // Med. Pregl. — 1989. — Vol. 42(9–10). — P. 332–334.
158. Streubel B., Vinatzer U., Lamprecht A. et al. T(3;14)(p14.1;q32) involving IGH and FOXP1 is a novel recurrent chromosomal aberration in MALT lymphoma // Leukemia. — 2005. — Vol. 19(4). — P. 652–658.
159. Suga S., Tanaka R., Tabata N. et al. Successful bone marrow transplantation in a child with combined IgG subclass deficiency and neutropenia // Bone Marrow Transplant. — 1995. — Vol. 16(6). — P. 847–848.
160. Suyama K., Kawasaki Y., Abe Y. et al. Development of common variable immunodeficiency in IgA– and IgG2-deficient patients with systemic lupus erythematosus // Pediatr. Nephrol. — 2012. — Vol. 27(3). — P. 489–492.
161. Take H., Kubota K., Tamura K. et al. Acquired total immunoglobulin G deficiency in a patient with primary biliary cirrhosis // J. Intern. Med. — 1995. — Vol. 238(3). — P. 289–292.
162. Tamura A., Agematsu K., Urasawa R. et al. Cardiac tamponade due to systemic lupus erythematosus in a 7-year-old boy with selective IgG subclass deficiency // Eur. J. Pediatr. — 1998. — Vol. 157(6) — P. 475–478.
163. Tashita H., Fukao T., Kaneko H. et al. Molecular basis of selective IgG2 deficiency. The mutated membrane-bound form of gamma2 heavy chain caused complete IGG2 deficiency in two Japanese siblings // J. Clin. Invest. — 1998. — Vol. 101(3). — P. 677–681.
164. Terada T., Kaneko H., Li A.L. et al. Analysis of Ig subclass deficiency: First reported case of IgG2, IgG4, and IgA deficiency caused by deletion of C alpha 1, psi C gamma, C gamma 2, C gamma 4, and C epsilon in a Mongoloid patient // J. Allergy Clin. Immunol. — 2001. – Vol. 108(4). — P. 602–606.
165. Tezcan I., Ersoy F., Sanal O. et al. IgG subclass deficiency in children with recurrent infections // Turk. J. Pediatr. — 1991. — Vol. 33(3). — P. 163–166.
166. Thakar Y.S., Chande C., Dhanvijay A.G. et al. Analysis of immunoglobulin deficiency cases: a five year study // Indian. J. Pathol. Microbiol. — 1997. — Vol. 40(3). — P. 309–313.
167. Tran Khai Hoan N., Karmochkine M., Laccourreye O., Bonfils P. Nasal polyposis and immunoglobulin-G subclass deficiency // Eur. Ann. Otorhinolaryngol. Head Neck. Dis. — 2014. — Vol. 131(3). — P. 171–175.
168. Van Kessel D.A., Horikx P.E., Van Houte A.J. et al. Clinical and immunological evaluation of patients with mild IgG1 deficiency // Clin. Exp. Immunol. — 1999. — Vol. 118(1). — P. 102–107.
169. Vanlerberghe L., Joniau S., Jorissen M. The prevalence of humoral immunodeficiency in refractory rhinosinusitis: a retrospective analysis // B-ENT. — 2006. — Vol. 2(4). — P. 161–166.
170. Vehapoglu A., Ozgurhan G., Demir A.D. et al. Recurrent meningitis in a child with IgG3 subclass deficiency // J. Pak. Med. Assoc. — 2014. — Vol. 64(8). — P. 963–965.
171. Vendrell M., de Gracia J., Rodrigo M.J. et al. Antibody production deficiency with normal IgG levels in bronchiectasis of unknown etiology // Chest. — 2005. — Vol. 127(1). — P. 197–204.
172. Venuta A., Penza R., Cellini M. et al. Primary hypergammaglobulinemic purpura associated with IgG2 deficiency. A case report // Clin. Exp. Rheumatol. — 1995. — Vol. 13(5). — P. 663–665.
173. Villiger P.M., Fey M.F., Lotz M., Tobler A. IgG2 and IgG4 subclass deficiency and Evan’s syndrome in an adult patient // J. Allergy Clin. Immunol. — 1992. — Vol. 90(4Pt1). — P. 693–694.
174. Vince N., Boutboul D., Mouillot G. et al. Defects in the CD19 complex predispose to glomerulonephritis, as well as IgG1 subclass deficiency // J. Allergy Clin. Immunol. — 2011. — Vol. 127(2). — P. 538–541.
175. Visitsunthorn N., Hengcrawit W., Jirapongsananuruk O., Luangwedchakam V. Immunoglobulin G (IgG) subclass deficiency in Thai children // Asian Pac. J. Allergy Immunol. — 2011. — Vol. 29(4). — P. 332–337.
176. Wågström P., Bengnér M., Dahle C. et al. Does the frequency of respiratory tract infections help to identify humoral immunodeficiencies in a primary health-care cohort? // Infect. Dis. (Lond). — 2015. — Vol. 47(1). — P. 13–19.
177. Wakefield D., Lloyd A., Brockman A. et al. Immunoglobulin subclass abnormalities in patients with chronic fatigue syndrome // Pediatr. Infect. Dis. J. — 1990. — Vol. 9(8). — S. 50–53.
178. Wilson N.W., Daaboul J., Bastian J.F. et al. Association of autoimmunity with IgG2 and IgG4 subclass deficiency in a growth hormone–deficient child // J. Clin. Immunol. — 1990. — Vol. 10(6). — P. 330–334.
179. Zenone T., Souquet P.J., Cunningham-Rundles C., Bernard J.P. Hodgkin’s disease associated with IgA and IgG subclass deficiency // J. Intern. Med. — 1996. — Vol. 240(2). — P. 99–102.
180. Zhao Y., Pan-Hammarström Q., Zhao Z. et al. Selective IgG2 deficiency due to a point mutation causing abnormal splicing of the Cgamma2 gene // Int. Immunol. — 2005. — Vol. 17(1). — P. 95–101.