
Журнал «» 2 (40) 2015
Вернуться к номеру
Відкрите рандомізоване порівняльне паралельне клінічне дослідження антигіпертензивної ефективності генеричного та брендового комбінованих препаратів ірбесартану та гідрохлортіазиду при гіпертонічній хворобі II–III ступеня
Авторы: Сіренко Ю.М., Радченко Г.Д., Поліщук С.А., Сідоренко П.І. — ДУ «ННЦ «Інститут кардіології ім. акад. М.Д. Стражеска» НАМН України, м. Київ
Рубрики: Кардиология
Разделы: Клинические исследования
Версия для печати
Статья опубликована на с. 70-81
На сьогодні артеріальна гіпертензія (АГ) є найбільш поширеним хронічним захворюванням у світі та в Україні [1]. Підвищений артеріальний тиск (АТ) спостерігається в кожної третьої дорослої людини. Поряд із високою поширеністю хронічно підвищений АТ є фактором ризику виникнення тяжких ускладнень (інфаркт міокарда, інсульт, серцева та ниркова недостатність, ретинопатія та ін.) та смерті. Ризик розвитку цих ускладнень збільшується при підвищенні як систолічного АТ (САТ), так і діастолічного АТ (ДАТ) у хворих усіх вікових категорій і статей [5, 6, 19]. Приблизні підрахунки показують, що прямі економічні збитки внаслідок тимчасової непрацездатності, інвалідності та передчасної смерті в Україні щороку перевищують 2 млрд гривень [1]. Крім того, певні економічні втрати для держави становлять витрати, пов’язані з лікуванням і реабілітацією зазначеної категорії хворих.
Особливістю АГ є і те, що дане захворювання відносно легко лікується постійним прийомом антигіпертензивних препаратів. Доведено, що зниження АТ лише на 10/5 мм рт.ст. зменшує ризик виникнення будь-яких серцево-судинних ускладнень на 10 %, а на 20/10 мм рт.ст. — до 20 % [5, 21, 23]. За даними клінічних досліджень, показано, що раціональне застосування антигіпертензивних препаратів дозволяє досягнути цільового АТ (менше 140 та 90 мм рт.ст.) майже у 80 % хворих. Проте реально тільки 15–20 % осіб із підвищеним АТ ефективно лікується [2, 8, 9, 11, 12, 16].
Зараз для медикаментозного лікування пацієнтів з АГ широко застосовуються препарати — блокатори ренін-ангіотензинової системи (РАС). Вони перш за все показані хворим з АГ та ознаками серцевої недостатності, ураженням нирок, метаболічним синдромом, цукровим діабетом, фібриляцією передсердь, гіпертрофією лівого шлуночка [4–6, 13, 23]. До цієї групи належать два класи ліків першої лінії терапії АГ — інгібітори ангіотензинперетворюючого ферменту (АПФ) та блокатори рецепторів ангіотензину ІІ (БРА). Найбільш популярними є їх комбінації з діуретиками, частіше з гідрохлортіазидом. У великих клінічних дослідженнях для лікування АГ частіше застосовувалася саме така комбінація.
Окрім спрощення режиму прийому антигіпертензивних препаратів, для підвищення прихильності хворого до терапії має значення ефективне зниження АТ при найменшій кількості побічних реакцій. У такому випадку існує більша вірогідність, що пацієнт буде дотримуватися призначеної йому терапії. Щодо ефективності, то на даний час жодна група антигіпертензивних препаратів не має переваг у ступені й частоті зниження АТ. Лідерами ж щодо найменшої кількості побічних реакцій серед антигіпертензивних препаратів є БРА. За даними багатьох досліджень, частота виникнення побічних реакцій при застосуванні препаратів цієї групи менша, ніж при прийомі плацебо [13, 21]. За даними K. Chen зі співавторами, призначення БРА асоціювалося з ймовірністю того, що пацієнти будуть продовжувати терапію у 81,9 % випадків, тоді як для діуретиків цей показник становив 70,9 %, інгібіторів АПФ — 74 %, антагоністів кальцію — 79 %, бета-адреноблокаторів — 76,7 % [5]. Тобто дана група ліків може забезпечувати більшу прихильність до лікування при тій же ефективності.
За механізмом дії БРА II принципово відрізняються від інгібіторів АПФ, оскільки рівень ангіотензину II при цьому в крові не змінюється, а іноді навіть підвищується. Крім того, при застосуванні інгібіторів АПФ, незважаючи на блокування утворення ангіотензину II, з часом рівень цього пептиду може повернутися до норми через альтернативні шляхи його утворення за допомогою інших ферментів, що каталізують перетворення ангіотензину I в ангіотензин II [24]. Крім того, як свідчить ряд експериментальних робіт, стимуляція ангіотензином II незаблокованих рецепторів може відіграти додаткову позитивну роль, оскільки відбувається збільшення NO як через брадикінін-залежний, так і брадикінін-незалежний механізми.
До представників БРА належить ірбесартан, ефективність та безпечність якого доведені у хворих на АГ, цукровий діабет із нефропатією, при метаболічному синдромі [7, 10, 14, 15, 17, 18, 20, 22].
Метою даного дослідження було оцінити терапевтичну еквівалентність за показниками ефективності генеричного комбінованого препарату ірбесартан/гідрохлортіазид (Ірбетан-Н, виробництва ПАО «Київський вітамінний завод») лікарському препарату Коапровель® 300/12,5 мг виробництва компанії Sanofi Winthrop Industri (Франція) в лікуванні гіпертонічної хвороби ІІ–ІІІ ступеня.
Завданнями дослідження були: вивчити терапевтичну ефективність препаратів, що досліджувалися, у лікуванні гіпертонічної хвороби; вивчити переносимість та можливі побічні реакції препаратів, що досліджувалися; порівняти ефективність лікування в основній та контрольній групах та оцінити терапевтичну еквівалентність препаратів, що досліджувалися.
Матеріали та методи
У дослідження було включено 90 пацієнтів з АГ II–III ступеня за класифікацією ВООЗ, які проходили стаціонарне або амбулаторне лікування у відділенні симптоматичних артеріальних гіпертензій Національного наукового центру «Інститут кардіології імені академіка М.Д. Стражеска» АМН України. Критеріями включення в дослідження були: вік 25–75 років, есенціальна АГ ІІ стадії, рівень систолічного та/або діастолічного АТ > 160/100 мм рт.ст. у кінці періоду відміни всіх антигіпертензивних препаратів, відсутність критеріїв виключення, підписання пацієнтом інформованої згоди на участь у дослідженні, можливості пацієнта до адекватного співробітництва. Не включалися пацієнти з рівнем АТ > 220/140 мм рт.ст.; вторинною та злоякісною АГ; з наявністю в анамнезі таких ускладнень, як інфаркт міокарда, інвазивні та неінвазивні втручання на серці, порушення мозкового кровообігу в строках менше ніж за три місяці до включення в дослідження; порушенням серцевого ритму (постійна форма фібриляції передсердь, часта шлуночкова або суправентрикулярна екстрасистолія, атріовентрикулярна блокада ІІ та ІІІ ступеня, синдром слабкості синусового вузла, брадикардія); серцевою недостатністю ІІІ–IV функціонального класу (NYHA); стенокардією напруження ІІІ–IV функціонального класу; тромбоемболією легеневої артерії; недостатністю лактази, галактоземією або синдромом порушення всмоктування глюкози/галактози в анамнезі; з анафілактичними реакціями в анамнезі; ретинопатією ІІІ та IV стадії, вираженою хронічною нирковою недостатністю (креатинін > 220 мкмоль/л, анурія); стенозом ниркових артерій, стенозами мітрального та аортального клапанів; із кардіоміопатіями; з декомпенсованим цукровим діабетом; із декомпенсованим ураженням печінки; порушенням кровотворення; з подагрою; з психічними розладами; з онкологічними захворюваннями; вагітні жінки або в період лактації; з підвищеною чутливістю до компонентів терапії в анамнезі; з анеміями; з інфекційними захворюваннями; з відсутністю контактної інформації; з необхідністю призначати інші антигіпертензивні або антиангінальні препарати; з епізодами гіпотензії; з клінічно значущими відхиленнями лабораторних показників, у тому числі з рівнями калію, натрію, сечової кислоти та кальцію сироватки крові, що виходили за межі норми; пацієнти, які беруть участь в інших клінічних дослідженнях.
Умовами припинення дослідження були: індивідуальна непереносимість препарату, поява тяжких або неочікуваних побічних реакцій, що потребують, на думку дослідника або пацієнта, відміни, значне погіршення загального стану в період дослідження, порушення пацієнтом протоколу дослідження, зміни лабораторних показників, що свідчать про потенційну загрозу для пацієнта, відкликання пацієнтом інформованої згоди. При достроковому припиненні участі в дослідженні пацієнт замінювався іншим згідно з критеріями включення та виключення. Новому пацієнту присвоювали наступний порядковий номер, і він включався у ту групу, з якої вибув пацієнт.
Дане клінічне дослідження проводилося відповідно до Закону України «Про клінічні засоби» та за етичними принципами, що викладені в Гельсінській декларації, а також згідно з «Інструкцією з проведення клінічних досліджень та експертизою матеріалів при клінічних дослідженнях», що затверджена Наказом МОЗ України від 01.11.2000 року. Дослідження було розпочато після затвердження протоколу дослідження комісією з питань етики при ДУ «ННЦ «Інститут кардіології імені академіка М.Д. Стражеска» НАМН України 08.04.2012.
Згідно з протоколом усім пацієнтами були проведені такі дослідження: збір анамнезу, вимірювання маси тіла та зросту, об’єктивне обстеження, вимірювання офісного САТ та ДАТ, частоти серцевих скорочень (ЧСС), біохімічне дослідження крові, загальноклінічні дослідження крові та сечі, тест на вагітність для жінок фертильного віку, електрокардіографія (ЕКГ), добове моніторування АТ (ДМАТ), огляд окуліста, визначення вираженості клінічних симптомів, реєстрація побічних явищ, оцінка ефективності лікування, контроль за виконанням режиму прийому препаратів (табл. 1).
/72/72.jpg)
Вимірювання АТ проводили ртутним сфігмоманометром вранці між восьмою та десятою годинами на всіх етапах дослідження (табл. 1). Реєстрацію САТ та ДАТ у положенні сидячи проводили на одній і тій же руці два рази з інтервалом у дві хвилини, якщо величини АТ не відрізнялися більше як на 5 мм рт.ст. [21]. При виявленні більшої різниці між отриманими величинами проводили третє вимірювання та обчислювали середнє значення з двох або трьох послідовних вимірювань. ЧСС визначали після другого вимірювання.
Реєстрацію електрокардіограми до, на 6-му тижні та в кінці лікування проводили на шестиканальному самописці «Юнікард» (Україна). Визначали наявність загальноприйнятих ознак гіпертрофії лівого шлуночка (індекс Соколова, індекс Корнелла, тривалість Корнелла) та порушення серцевого ритму.
ДМАТ проводили на портативному апараті АВРМ-04 («Метідек», Угорщина) в кінці періоду вимивання, на 6-му тижні та в кінці періоду лікування. При цьому вивчали такі показники: середньодобовий (т), денний (д), нічний (н) та максимальний САТ, середньодобовий, денний, нічний та максимальний ДАТ, ЧСС [25]. Також визначали індекс варіабельності (Ст.від.) [25]. Крім того, за допомогою програмного забезпечення апарата вираховували добовий індекс (ДІ) — відсоток зниження нічного АТ порівняно з денним. Моніторування проводили в такому режимі: у денний час — кожні 15 хвилин, уночі (з 22.00 до 6.00) — кожні 30 хвилин. Хворі вели звичайний спосіб життя, з побутовими фізичними і психоемоційними навантаженнями [25].
Біохімічні аналізи виконувалися на автоматичному біохімічному аналізаторі А-25 (Франція) в ДУ «ННЦ «Інститут кардіології імені академіка М.Д. Стражеска» НАМН України до та в кінці дослідження. Визначали рівень білірубіну, креатиніну, калію, натрію, тригліцеридів, загального білка, АЛТ, АСТ, сечової кислоти, глюкози та холестерину сироватки крові. Тест на вагітність проводили жінкам фертильного віку за допомогою смужкового тесту.
Оцінку ефективності лікування проводили на основі динаміки АТ (офісного та добового), а також суб’єктивних скарг хворого. Головними критеріями ефективності були: зниження офісного САТ не менше ніж на 20 мм рт.ст. та ДАТ не менше ніж на 10 мм рт.ст. або досягнення цільового рівня САТ/ДАТ (менше 140/90 мм рт.ст.). Вторинними критеріями ефективності були: зниження середніх значень САТ і ДАТ за 24 години, у період сну та в денний період. Переносимість лікарського препарату оцінювали за шкалою:
— добра (при об’єктивному обстеженні в динаміці не виявляються жодні патологічні зміни або клінічно значущі відхилення, дані лабораторного обстеження вірогідно не змінюються і не виходять за межі норми, пацієнт не відмічає побічних реакцій);
— задовільна (при обстеженні в динаміці виявляються незначні зміни, що мають транзиторний характер та не потребують додаткових медичних втручань або зміни схеми лікування, дані лабораторних досліджень відхиляються від норми незначно, спостерігаються незначні побічні явища, що не викликають у пацієнта серйозних проблем та не потребують відміни препарату);
— незадовільна (при обстеженні в динаміці виявляються патологічні зміни, що потребують відміни препарату та проведення додаткових медичних втручань, дані лабораторних обстежень є клінічно значущими та потребують додаткового обстеження, має місце небажана побічна дія, що дуже негативно впливає на стан пацієнта та потребує відміни препарату і проведення додаткових медичних втручань.
Враховували та оцінювали у балах ступінь вираженості таких ознак (суб’єктивних скарг): головний біль, запаморочення, порушення сну, галюцинації/депресія, слабкість м’язів, диспептичні явища (діарея, запор, нудота, болі в животі), задишка, ортостатична гіпотензія, порушення зору, набряки та інші, вказані в індивідуальній карті пацієнта. Ступінь вираженості скарг визначався за бальною системою: 0 — відсутність скарг, 1 — незначна вираженість скарг, 2 — помірна вираженість, 3 — значна вираженість.
Усі дослідження проводилися після 3-денної відміни антигіпертензивних препаратів, що пацієнт отримував до включення в дослідження, та через місяць лікування. Контроль офісного АТ та реєстрація побічних явищ проводилися на кожному етапі обстеження.
До початку дослідження всім пацієнтам відміняли на 3 дні антигіпертензивну терапію, після чого проводили лабораторно-інструментальні дослідження. Тривалість періоду відміни була визначена, виходячи з того, що більшість препаратів мають період напіввиведення 12–24 години і вони повністю зникають з крові протягом у середньому 48–72 годин. При підвищенні АТ у період відміни медична допомога надавалася пацієнту згідно з п. 16 Наказу № 24 від 17.01.05 «Про затвердження протоколів надання медичної допомоги за спеціальністю «Медицина невідкладних станів».
Пацієнтам основної групи (n = 45) призначали препарат Ірбетан-Н, таблетки, виробництва ПАТ «Київський вітамінний завод». Препарат призначався по 1 таблетці (300 мг ірбесартану + 12,5 мг гідрохлортіазиду) один раз на добу вранці. Курс лікування становив 28 днів. Пацієнт приймав препарат в один і той же час. Таблетки приймали цілими та запивали невеликою кількістю рідини під час або одразу після прийому їжі. Курс прийому становив 28 днів. Пацієнти контрольної групи (n = 45) отримували за аналогічною схемою зареєстрований в Україні референтний препарат Коапровель® (300 мг ірбесартану + 12,5 мг гідрохлортіазиду) виробництва фірми Sanofi Winthrop Industrie (Франція).
Під час дослідження не дозволявся прийом інших антигіпертензивних препаратів, великої кількості рідини, нестероїдних та стероїдних протизапальних препаратів, симпатоміметичних препаратів, індукторів мікросомального окислення. Під час кожного візиту проводився контроль прийому ліків, підраховували кількість прийнятих пацієнтом таблеток та кількість днів, що минули від попереднього візиту. Якщо коефіцієнт прихильності до лікування (кількість випитих таблеток/кількість днів прийому) був меншим за 0,75, пацієнт виключався з дослідження.
Статистичну обробку результатів проводили після створення баз даних у системах Microsoft Excel. Середні показники обстежених пацієнтів визначали за допомогою пакета аналізу в системі Microsoft Excel. Усі інші статистичні розрахунки проводили за допомогою програми SPSS 13.0. Нормальність рядів визначалася за допомогою критерію Шапіро — Уїлка. При нормальному розподілі вірогідність різниці середніх на етапах лікування визначалася за допомогою парного двовибіркового тесту, вірогідність різниці між групами — за допомогою незалежного t-тесту для середніх після визначення характеру розподілу показників. Ефективність у групах та різниця у групах за розподілом наявності тої або іншої ознаки оцінювалася за дихометричною змінною за допомогою критерію x2.
Результати
У дослідження було рандомізовано 90 пацієнтів із II та III ступенем підвищення АТ. Ще 3 пацієнтів було виключено на етапі до рандомізації — у них відмічалося значне підвищення АТ у період вимивання, що потребувало призначення комбінованої антигіпертензивної терапії. Побічні явища виникли у 3 (6,7 %) пацієнтів основної групи та у 2 (4,4 %) — контрольної групи. Проте жодного пацієнта після рандомізації з дослідження виключено не було. В аналіз кінцевої ефективності терапії було включено по 45 пацієнтів основної та контрольної груп. Загальна клініко-демографічна характеристика пацієнтів, включених у дослідження, наведена в табл. 2.
/74/74.jpg)
Як видно з табл. 2, у дослідженні брала участь майже однакова кількість жінок та чоловіків, які в більшості своїй мали досвід прийому інгібіторів АПФ, середнього віку 53,4 ± 2,5 року, які мали переважно надмірну масу тіла та порушення ліпідного обміну. Окрім того, 11,1 % включених у дослідження пацієнтів мали ішемічну хворобу серця та 22,2 % — порушення вуглеводного обміну (підвищений рівень глюкози натще, цукровий діабет, знижена толерантність до глюкози при проведенні глюкозотолерантного тесту). Середній рівень АТ у кінці періоду вимивання відповідав критеріям включення. Середньодобові рівні САТ і ДАТ також були підвищеними відповідно до ступеня АГ.
Пацієнтам обох груп було проведено фізикальне обстеження, офісне вимірювання АТ, ДМАТ, ЕКГ. Клініко-демографічна характеристика рандомізованих груп пацієнтів наведена в табл. 3. Як видно з табл. 3, на початку дослідження групи пацієнтів, що порівнювалися, вірогідно не відрізнялися за основними клініко-демографічними показниками.
/75/75_2.jpg)
В обох групах переважали хворі з надмірною масою тіла, підвищеними рівнями загального холестерину та тригліцеридів сироватки крові. В обох групах 11,1 % пацієнтів мали ішемічну хворобу серця та в понад 20 % пацієнтів відмічалося порушення обміну глюкози (або цукровий діабет, або порушення толерантності до глюкози, або підвищений рівень глюкози натще). Тобто як мінімум кожен п’ятий пацієнт зараховувався до групи дуже високого ризику виникнення серцево-судинних ускладнень. Групи вірогідно не відрізнялися за рівнем АТ.
/75/75.jpg)
Під впливом призначеної додаткової терапії спостерігалося вірогідне зниження рівнів офісного САТ, ДАТ та ЧСС в обох групах. Ці дані наведені на рис. 2 та в табл. 4. Як видно з них, у середньому після рандомізації офісний САТ знизився з 176,2 ± 2,5 мм рт.ст. до 139,9 ± 2,5 мм рт.ст. (Р < 0,001) у 1-й групі та з 180,1 ± 2,1 мм рт.ст. до 139,9 ± 2,2 мм рт.ст. (Р < 0,001) у 2-й групі. Рівень ДАТ відповідно знизився з 106,1 ± 1,8 мм рт.ст. до 83,7 ± 1,2 мм рт.ст. (Р < 0,001) та з 107,1 ± 1,4 мм рт.ст. до 82,3±1,4 мм рт.ст. (Р < 0,001). При цьому вірогідне зниження офісного САТ/ДАТ і в контрольній групі, і в основній відбувалося вже на 7-й день прийому препарату. За ступенем зниження офісного САТ і ДАТ контрольний препарат був незначно, але вірогідно більш ефективний на етапах лікування. Проте це пояснювалося більш високим рівнем АТ у групі порівняння на початку дослідження, адже за рівнем досягнутого АТ групи вірогідно не відрізнялися на відповідних етапах лікування.
/76/76.jpg)
Офісна ЧСС вірогідно зменшилася в обох групах із 77,2 ± 2,3 уд/хв та 79,3 ± 2,4 уд/хв на початку дослідження та 70,5 ± 1,9 уд/хв та 70,9 ± 1,9 уд/хв у кінці дослідження відповідно в першій та другій групах (Р < 0,05 та 0,01 відповідно). За ступенем зменшення ЧСС групи вірогідно не відрізнялися. Зниження ЧСС у нашому дослідженні підтверджує дані літератури щодо антисимпатичної дії блокаторів ренін-ангіотензинової системи.
/76/76_2.jpg)
Загалом цільового офісного АТ було досягнуто у 38 (84,4 %) та 40 (88,9 %) пацієнтів на етапі 4 тижнів лікування відповідно в основній та контрольній групах (Р = НВ). Частка хворих, у яких офісний САТ/ДАТ вірогідно знизився на > 20/10 мм рт.ст., становила відповідно в першій та другій групі 95,6 % (n = 43) та 97,8 % (n = 44), P = НВ. Тобто обидва препарати були однаково ефективними щодо зниження офісного АТ при 4-тижневому прийомі. Ступінь зниження АТ був порівнянний із даними літератури для комбінованих препаратів [7, 17, 18, 20, 22].
Результати ДМАТ наведено в табл. 5. Як видно з табл. 5, на початку дослідження, як і слід було очікувати, тСАТ і тДАТ були меншими, ніж офісні показники, і групи вірогідно не відрізнялися між собою як за показниками на початку дослідження, так і на етапі рандомізації. На фоні призначеного лікування спостерігалося вірогідне зниження середньодобових САТ/ДАТ, денних САТ/ДАТ та нічних САТ/ДАТ в обох групах. Ступінь зниження середньодобових САТ/ДАТ, денних САТ/ДАТ та нічних САТ/ДАТ становив 19,3/14,5; 21,3/15,3; 18,2/8,8 та 20,4/16,1; 23,6/18,3 і 22,8/12,4 мм рт.ст. відповідно в першій та другій групах (Р = 0,05/0,001; Р = НВ/0,005; Р = 0,02/0,02).
/77/77.jpg)
/78/78.jpg)
Як видно, у другій групі призначений референтний препарат був більш ефективним щодо ступеня зниження АТ, виміряного при добовому моніторуванні. Проте це можна пояснити дещо більшим рівнем САТ і ДАТ на початку дослідження у другій групі, адже за досягнутими рівнями САТ і ДАТ групи вірогідно не відрізнялися. Варіабельність дСАТ/ДАТ та нСАТ вірогідно знизилася в обох групах. Проте ступінь зниження нДАТ за рахунок більш високих початкових показників був вірогідно більший у групі референтного препарату. За даними літератури, зменшення варіабельності АТ можна вважати позитивною рисою лікування, адже підвищена варіабельність асоціюється з більшою частотою виникнення несприятливих подій [25]. Середній ДІ на початку дослідження був у межах норми в обох групах. На фоні лікування він вірогідно зменшився у першій групі, але в межах нормальних показників і залишався більш низьким, ніж у групі референтного препарату. За величиною ДІ частка хворих, які характеризувалися як нон-дипери, становила 15 (33,3 %) та 11 (24,4 %) осіб відповідно в першій та другій групах (Р = НВ). На фоні лікування вона зменшилася, але невірогідно в обох групах і становила відповідно 12 (26,7 %) та 10 (22,2 %) осіб, Р = НВ. Загалом цільового середньодобового АТ (< 130/80 мм рт.ст.) було досягнуто у 25 (55,6 %) пацієнтів 1-ї групи та у 27 (60,0 %) пацієнтів 2-ї групи (різниця невірогідна). Менший відсоток пацієнтів, які досягли цільового АТ за даними ДМАТ, є цілком закономірним. Адже з даних літератури відомо, що ефективність антигіпертензивної терапії значно менша, якщо її оцінювати за допомогою методу ДМАТ порівняно з офісним вимірюванням [25]. Показники тЧСС, дЧСС та нЧСС вірогідно та більшою мірою знизилися лише у другій групі.
У нашому дослідженні не спостерігалося вірогідної зміни показників ЕКГ (табл. 6). Ми не відмітили вірогідної зміни загальноклінічних та біохімічних показників, що вивчалися (табл. 7), що свідчило про метаболічну нейтральність даних препаратів. В обох групах спостерігалася тенденція до зменшення рівня загального холестерину та глюкози в обох групах.
Ми не спостерігали значних та вірогідних змін в аналізі сечі в жодній групі обстежених пацієнтів (табл. 8).
Розподіл пацієнтів залежно від лабораторних показників (із відхиленням від норми та без них) до та в кінці лікування наведено в табл. 9. Як видно з табл. 9, на фоні лікування в обох групах вірогідно не змінилися кількість та частка пацієнтів, які мали відхилення лабораторних показників. Спостерігалася тенденція до зменшення частки пацієнтів із відхиленням рівня глюкози сироватки крові. Групи вірогідно не відрізнялися за часткою пацієнтів із відхиленнями лабораторних показників між собою на етапах лікування.
/79/79.jpg)
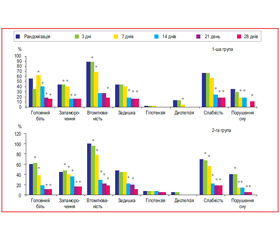
Динаміка вираженості суб’єктивних скарг хворих у групах наведена в табл. 10. Як видно з табл. 10, в обох групах вірогідно зменшилася вираженість таких симптомів, як головний біль (перш за все за рахунок зменшення АТ), запаморочення, підвищена втомлюваність, задишка, нудота (за рахунок зниження АТ та зменшення піків підйому АТ), слабкість та порушення сну. Розподіл пацієнтів залежно від наявності в них скарг на етапах лікування наведено на рис. 3. Як видно з рис. 3, лікування і препаратом Ірбетан-Н, і Коапровель® сприяло значному та вірогідному зменшенню частки пацієнтів із наявністю скарг. Групи вірогідно не відрізнялися між собою за частотою, з якою зустрічалася та або інша скарга в обстежених пацієнтів, та суттєво не відрізнялися за динамікою зміни частоти, з якою зустрічалися скарги.
/80/80.jpg)
Оцінка переносимості препаратів, що порівнювалися, наведена на рис. 4. Як видно з рис. 4, 43 (95,5 %) пацієнти першої групи та 44 (97,8 %) другої групи добре або задовільно переносили призначене лікування — з них 38 (84,4 %) та 39 (86,7 %) пацієнтів відповідно першої та другої групи мали добру переносимість.
Побічні явища виникли у 3 (6,7 %) пацієнтів основної групи та у 2 (4,4 %) — контрольної групи (Р = НВ). У 1-й групі у всіх 3 пацієнтів (6,7 %) був зареєстрований головний біль, що, імовірно, був пов’язаний із недостатнім контролем АТ у перші дні лікування основним препаратом (пацієнт К.І.П., № 21, 49 років, пацієнтка М.В.А., № 27, 61 року, пацієнтка М.Ю.І., № 29, 53 років). У другій групі в 1 пацієнтки виник головний біль (пацієнтка С.О.В., № 44, 50 років) і в 1 пацієнта спостерігалося запаморочення (пацієнт П.В.П., № 30, 47 років). Жоден включений у дослідження пацієнт не потребував відміни антигіпертензивних препаратів, що призначалися.
Таким чином, частота виникнення побічних реакцій в обох групах вірогідно не відрізнялася між собою і була порівнянна з даними літератури [17, 18, 20, 22].
Як указувалося вище, цільового офісного АТ було досягнуто у 38 (84,4 %) та 40 (88,9 %) пацієнтів на етапі 4 тижнів лікування відповідно в основній та контрольній групах (Р = НВ). Частка хворих, у яких офісний САТ/ДАТ вірогідно знизився на > 20/10 мм рт.ст., становила в першій та другій групі відповідно 95,6 % (n = 43) та 97,8 % (n = 44), P = НВ. Тобто первинна кінцева точка була досягнута в переважної більшості пацієнтів, включених у дослідження. Таким чином, висока ефективність була відзначена у 95,6 та 97,8 % хворих (Р = НВ) (рис. 5).
Таким чином, застосування препарату Ірбетан-Н, таблетки, виробництва ПАТ «Київський вітамінний завод» ефективно знижувало АТ, виміряний як в офісі, так і при ДМАТ. Препарат, що досліджувався, вірогідно не відрізнявся за своєю антигіпертензивною ефективністю від препарату порівняння Коапровель®, таблетки, виробництва компанії Sanofi Winthrop Industrie (Франція). Зниження АТ на фоні прийому обох препаратів супроводжувалося вірогідним зменшенням варіабельності АТ. Терапія обома препаратами була метаболічно нейтральною та добре переносилась хворими.
Висновки
1. Препарат Ірбетан-Н виробництва ПАТ «Київський вітамінний завод» (Україна) характеризувався вірогідним та ефективним зниженням рівнів офісного САТ, ДАТ відповідно на 36,3 і 22,4 мм рт.ст., що було еквівалентно зниженню офісного САТ і ДАТ на фоні лікування препаратом Коапровель® виробництва компанії Sanofi Winthrop Industrie (Франція) — 40,2 і 25,8 мм рт.ст. За рівнем досягнутого АТ групи вірогідно не відрізнялися. Цільового офісного АТ було досягнуто у 84,4 % пацієнтів, які отримували препарат Коапровель®. Частка хворих, у яких офісний САТ/ДАТ вірогідно знизився на > 20/10 мм рт.ст., становила відповідно в першій та другій групі 95,6 та 97,8 % (P = НВ).
2. На фоні лікування в обох групах спостерігалося вірогідне еквівалентне зниження рівнів середньодобових, денних, нічних САТ/ДАТ відповідно у групі Ірбетану-Н на 9,3/14,5; 21,3/15,3, 18,2/8,8 мм рт.ст., а у групі Коапровелю® — на 20,4/16,1; 23,6/18,3 та 22,8/12,4 мм рт.ст. У кінці дослідження групи вірогідно не відрізнялися між собою за рівнем досягнутого АТ. Цільового середньодобового АТ (< 130/80 мм рт.ст.) було досягнуто у 55,6 % пацієнтів 1-ї групи та в 60,0 % пацієнтів 2-ї групи.
3. Висока антигіпертензивна ефективність препаратів, що порівнювалися, підтверджувалася вірогідним зменшенням підвищеної варіабельності денного та нічного САТ.
4. Обидва препарати характеризувалися низькою частотою виникнення побічних реакцій (6,7 % — у групі препарату Ірбетан-Н та 4,4 % — у групі Коапровель®), були метаболічно нейтральними та добре переносилися хворими. За переносимістю препарати вірогідно не відрізнялися між собою.
1. Артеріальна гіпертензія — медико-соціальна проблема / В.М. Коваленко, М.І. Лутай, Є.П. Свіщенко, Ю.М. Сіренко, І.П. Смірнова. — К.: Інститут кардіології ім. М.Д. Стражеска АМН України, 2002. — 102 с.
2. Горбась І.М. Контроль артеріальної гіпертензії серед населення: стан проблеми за даними епідеміологічних досліджень // Укр. кардіол. журнал. — 2007. — № 2. — С. 21–26.
3. Настанова та клінічний протокол надання медичної допомоги «Артеріальна гіпертензія». Наказ МОЗ України № 384 від 24.05.2012. — Київ, 2012. — 107 с.
4. Рекомендації Українського товариства кардіологів з профілактики та лікування артеріальної гіпертензії. — Київ: Віпол, 2008. — 83 с.
5. 2007 European Society of Hypertension — European Society of Cardiology guidelines for management of arterial hypertension // J. Hypertension. — 2007. — Vol. 25. — P. 1105–1187.
6. 2013 ESH/ESC Guidelines for the management of arterial hypertension TheTask Force for the management ofarterial hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC) // Journal of Hypertension. — 2013. — Vol. 31. — P. 1281–1357.
7. Arhofer K.G., Muenzel F., Krekler M. Effect of the Angiotensin Receptor Blocker Irbesartan on Metabolic Parameters in Clinical Practice: the DO-IT Prospective Observational Study // Cardiovasc. Diabetol. — 2007. — 6(1). — P. 36.
8. Basta E., Barkis.G. Сhoices and goals in the treatment of diabetic hypertensive patients // Curr. Hypert. Rep. — 2001. — Vol. 3. — P. 387–391.
9. Bonds D., Palla S., Bertoni A. et al. Hypertension Treatment in the Ambulatory Setting: comparison by race and gender in a National survey // J. Clin. Hypertens. — 2004. — Vol. 6. — P. 223–230.
10. Bramlage P., Durand-Zaleski I., Desai N., Pirk O., Hacker C. The value of irbesartan in the management of hypertension // Expert Opin. Pharmacother. — 2009. — Vol. 10. — P. 1817–31.
11. Burt V.L., Cutler J.A., Higgins M. et al. Тrends in the prevalence, awareness, treatment and control of hypertension in the adult US population. Data from the health examination surveys 1960–1991 // Hypertens. — 1995. — Vol. 26. — P. 60–65.
12. Chatellier G., Menard J., Devries C. et al. Blood pressure control in hypertension center // J. Hypertens. — 1987. — Vol. 5. — P. 47–49S.
13. Elliot H., Connel J., McInnes G. The Year in hypertension. — Clinical Publishing Services Ltd: Oxford, 2001. — 369 p.
14. Fogari R., Ambrosoli S., Corradi L. et al. 24-hour blood pressure control by once-daily administration of irbesartan assessed by ambulatory blood pressure monitoring: Irbesartan Multicenter Investigators’ Group // J. Hypertens. — 1997. — Vol. 15. — P. 1511–1518.
15. Havranek E.P., Thomas I., Smith W.B. et al. Dose-related beneficial long-term hemodynamic and clinical efficacy of irbesartan in heart failure // J. Am. Coll. Cardiol. — 1999. — Vol. 33. — P. 1174–1181.
16. Hypertension Primer. The essentials of high blood pressure. Basic science. Population science and clinical management. Fourth edition. From the council on high blood pressure research American Heart Association. — Lippincott Williams & Wilkins: Dallas, 2008. — 610 p.
17. Kintscher U., Bramlage P., Paar W.D. et al. Irbesartan for the treatment of hypertension in patients with the metabolic syndrome: a sub analysis of the Treat to Target post authorization survey. Prospective observational, two armed study in 14,200 patients // Cardiovasc. Diabetol. — 2007. — 6. — P. 12.
18. Lewis E.J., Hunsicker L.G., Clarke W.R. et al. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes // N. Engl. J. Med. — 2001. — 345. — P. 851–860.
19. Mancia G., Sega R., Milesi C. et al. Blood pressure control in the hypertensive population // Lancet. — 1997. — Vol. 348. — P. 454–457.
20. Massie B.M., Carson P.E., McMurray J.J. et al. Irbesartan in patients with heart failure and preserved ejection fraction // N. Engl. J. Med. — 2008. — Vol. 359. — P. 172.
21. Moser M. Clinical management of Hypertension. Fourth Edition. — Caddo: Professional Communications, Inc., 1999. — 240 p.
22. Parving H.H., Lehnert H., Brochner-Mortensen J. et al. The effect of irbesartan on the development of diabetic nephropathy in patients with type 2 diabetes // N. Engl. J. Med. — 2001. — 345. — P. 870–8.
23. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation and Treatment of High Blood Pressure. US Department of Health and Human Service. NIH Publication No. 03–5233, 2003. — 34 p.
24. Urata H., Healey B., Stewart R.W., Bumpus F.M., Husain A., Angiotensin II-Forming Pathways in Normal and Failing Human Hearts // Cirs. Res. –1990. — Vol. 66. — P. 883–90.
25. White W. Blood pressure monitoring in Cardiovascular Medicine and Therapeutics. — N. Jersy: Humana Press, 2001. — 308 p.