Статтю опубліковано на с. 14-20
Вступ
Поширеність атопічних захворювань (АЗ), таких як атопічний дерматит, алергічний риніт, бронхіальна астма, у розвинених країнах становить приблизно 20 % і невпинно зростає протягом останніх десятиліть [27]. Щорічний приріст атопічних захворювань у світі є глобальною медико-соціальною проблемою, що призвело до виникнення терміна «алергічна пандемія» [8].
Характерна послідовність виникнення клінічних проявів атопічних захворювань протягом дитячого віку, що пов’язана з розвитком алергічного запалення в шокових органах, має назву «атопічний марш» [26]. Загалом прояви атопічного дерматиту передують розвитку алергічного риніту та бронхіальної астми і вважаються початковим етапом «атопічного маршу» [2, 8, 26]. Так, розвиток алергічного запалення в шкірі призводить до появи великої кількості циркулюючих активованих еозинофілів, імуноглобулінів Е (IgE), Т-хелперів 2-го типу (Th2), що транслокуються до верхніх і нижніх дихальних шляхів і ініціюють запальний процес [4].
В основі патогенезу всіх атопічних захворювань лежить генетично детермінована змінена реактивність на антигени зовнішнього середовища з гіперпродукцією ІgE [1]. Спільні генетичні варіанти, таким чином, мають плейотропний ефект і є об’єктом активного вивчення, оскільки саме вони визначають перспективні напрямки терапії АЗ [9]. На сьогодні відомо більше ніж 300 генів, однонуклеотидні поліморфізми в яких асоційовані із розвитком атопічних захворювань [14, 22]. Для систематизації отриманих даних використовуються різні класифікації кандидатних генів. За однією з класифікацій, передбачається виділення 4 категорій генів, які визначають ризик розвитку атопічних захворювань на етапах антигенпрезентації (HLA-Pep-TCR), гіперпродукції IgE (IL-4Rα, IFN-γR1), синтезу та вивільнення медіаторів (LTC4S), та гени, які визначають орган-мішень (ADAM33, –IL-13R) [13]. Інші автори виділяють гени, спільні для всіх атопічних захворювань, гени, які кодують білки головного комплексу гістосумісності (MHC), інтерлейкіни (IL), високоафінні компоненти рецепторів до IgE та специфічні гени, характерні для певної нозології [9]. Останнім часом підкреслюється важливість вивчення асоціацій значущих генів із атопічними хворобами в кожній із популяцій, оскільки відмінності в розподілі алельних варіантів не дозволяють впроваджувати результати генетичних досліджень в практику охорони здоров’я в усьому світі [9].
Автофагія — процес підтримки внутрішньоклітинного гомеостазу шляхом деградації довгоживучих білків та органел для побудови необхідних макромолекул і формування таким чином адекватної відповіді клітини на стресові фактори [21, 30].
Виділяють принаймні три типи автофагії: мікроавтофагія, шаперон-опосередкована автофагія і макроавтофагія [21, 29]. Maкроавтофагія вважається основним шляхом деградації протеїнів. При цьому вміст цитоплазми, включаючи органели, наприклад мітохондрії, секвеструється ізоляційною мембраною, закриття країв якої призводить до утворення структури з подвійною мембраною, що має назву «автофагосома» [21]. У подальшому відбувається злиття автофагосоми з лізосомою, лізосомальні ензими при цьому деградують поглинені матеріали та внутрішню мембрану автофагосоми [19, 30].
Молекулярні процеси, що беруть участь у формуванні автофагосоми, складаються з декількох етапів: індукція, ініціювання формування та елонгації мембрани, злиття з лізоcoмою, при цьому було виділено принаймні 31 ген, пов’язаний з автофагією (ATG) [5, 7, 29]. Автофагія бере участь у розвитку алергічного запалення на всіх етапах, а гени, що кодують білки автофагії, відносяться до спільного генетичного базису атопічних захворювань.
Автофагія забезпечує презентацію цитозольних, ядерних антигенів, а також антигенів внутрішньоклітинних патогенів у МНС ІІ комплексі антигенпрезентуючих та МНС ІІ-позитивних епітеліальних клітин СD4+ Т-клітинам. Так, екзогенні антигени обробляються в автолізосомальних компартментах за допомогою протеаз та завантажуються на молекули МНС класу II, які транспортуються від ендоплазматичного ретикулуму до відсіків МНС ІІ типу (MIICs) [24]. При цьому МНС ІІ-позитивні АПК (В-клітини, моноцити, дендритні клітини) мають стабільно високі рівні автофагічної активності. Таким чином, можна зробити висновок, що активація Т-хелперів залежить від інтенсивності автофагії [25].
СD4+ Т-клітини та CD8+ T-клітини виявляють базальні рівні активності автофагії, які значно зростають при активації Т-клітинного рецептора або за наявності відповідних цитокінів. [24]. Так, при активації СD4+ Т-клітин відзначається різке зростання кількості автофагосом. Рівень автофагічної активності при цьому значно вищий у Тh2-клітини, що, можливо, пов’язано з високою активністю каспаз, які спричиняють апоптоз та одночасно інгібують автофагію у Тh1-клітини [17]. Окрім того, Тh2-клітини більш сприйнятливі до нестачі нутрієнтів, що є класичним активатором автофагії [12].
Цитокіновий профіль середовища має значний вплив на диференціацію Т-клітин. Так, –IL-12 призводить до дозрівання Th1-клітин-ефекторів та цитотоксичних CD8+-клітин, а перетворення в Тh2-клітини відбувається під впливом IL-4 [3]. Автофагія також знаходиться під контролем цитокінів. IFN-γ та TNF-α стимулюють автофагію в макрофагах, а IL-4 та IL-13, класичні цитокіни Тh2-клітини, навпаки, пригнічують [10, 15, 24].
Функціями цитокінів та хемокінів Тh2-клітини є інгібування клітинної імунної відповіді, стимуляція продукції IgE В-клітинами, міграція еозинофілів до осередку запалення. Зв’язування IgE з відповідними рецепторами на базофілах і мастоцитах призводить до вивільнення останніми гістаміну, серотоніну, еозинофільних хемотаксичних факторів, протеаз, простагландинів, лейкотрієнів тощо. Виникнення клінічних симптомів атопічних хвороб пов’язане саме з впливом даних медіаторів [1].
Автофагія відіграє важливу роль у розвитку та диференціації В-лімфоцитів, посилення експресії В-клітинного рецептору асоціюється з активацією автофагії [28]. B.C. Miller та ін. [18] встановили, що протеїн ATG5 є необхідним для підтримки життєздатності клітин-попередників В-клітин, підтримки загальної чисельності периферичних В-1-клітин. Також в іншому дослідженні було підтверджено значення автофагії у процесах дозрівання гранул мастоцитів, їх транслокації та зливанні з мембраною під час дегрануляції. Інгібування автофагії призводить до селективного пригнічення антигеніндукованої дегрануляції тучних клітин [20].
Цитокіновий профіль, а саме IL-13, рівень якого значно підвищується при бронхіальній астмі, також сприяє активації трансформуючого фактора росту β1. При цьому його взаємодія з активними формами кисню призводить до ремоделювання дихальних шляхів шляхом вивільнення колагену з фібробластів, стимуляції міграції фібробластів до субепітеліального шару та їх диференціаціх в міофібробласти [23]. Являючи собою потужний антиоксидантний механізм, автофагія відповідає за деградацію окислених протеїнів і пошкоджених органел, у тому числі деполяризованих мітохондрій. У патологічних умовах окисний стрес та дисбаланс активних форм кисню є частим ускладненням, що супроводжують хронічні запальні процеси. Окисний стрес активує автофагію через вивільнення асоційованих з ушкодженнями молекулярних патернів (DAMPs), до яких відносяться білки теплового шоку, родина IL-1, деякі компоненти плазми. Автофагія, у свою чергу, регулює транслокацію та вивільнення патернів, що зрештою визначає долю клітини [16]. Дослідження на мишах показали, що при інгібуванні автофагії порушується структура війчастого епітелію, у цитоплазмі епітеліальних клітин бронхіол присутні мішкоподібні та концентричні мембраноїдні структури, епітелій набряклий, виявлено гіперреактивніть дихальних шляхів при холінергічних стимулах [11].
Враховуючи вищенаведене, висловлено припущення, що однонуклеотидний поліморфізм rs510432 гена автофагії 5 (ATG5) може бути асоційований із розвитком атопічних захворювань у дітей. Для перевірки цієї гіпотези проведено дослідження відмінності в частоті алельних варіантів поліморфізму rs510432 гена ATG5 у дітей з проявами «атопічного маршу» та у здорових дітей без випадків атопічних захворювань в сімейному анамнезі. Окрім того, визначена ймовірність розвитку «атопічного маршу» у носіїв патологічної алелі та з’ясована наявність взаємозв’язку між алельним варіантом та ступенем тяжкості атопічних захворювань у дітей. Поліморфізм rs510432 гена ATG5 був обраний для генотипування, оскільки він зустрічається в європоїдів та гіпотетично впливає на розвиток та прогресування «атопічного маршу».
Матеріали та методи
У дослідження включено 98 дітей віком від 5 до 18 років, хворих на бронхіальну астму та атопічний дерматит, із них у 31 хворого діагностовано алергічний риніт. Усі пацієнти перебували на стаціонарному лікуванні в алергологічному відділенні Київської міської дитячої клінічної лікарні № 2. Усі хворі мали випадки атопічних захворювань у сімейному анамнезі. До групи контролю увійшло 98 здорових дітей віком 5–18 років без випадків атопічних захворювань у сімейному анамнезі. Проведені наступні методи дослідження: загальноклінічне та лабораторне обстеження, що включало загальний аналіз крові, загальний аналіз сечі, копрограму, загальний IgE, пікфлоуметрія, спірометрія, шкірні прик-тести, визначення поліморфізму rs510432 гена ATG5. Проведення дослідження погоджено з Комісією з питань етики Національного медичного університету імені О.О. Богомольця. Батьки підписали інформовану згоду на включення дітей в дослідження.
Виділення ДНК та полімеразна ланцюгова реакція
Виділення ДНК проводили із букального епітелію за допомогою реагентів DiatomTM Prep 200 («Лаборатория Изоген», РФ), згідно з рекомендаціями виробника. Концентрацію загального ДНК визначали за допомогою NanoDrop спектрофотометра ND-1000 (NanoDrop Technologies Inc., США). Реакції ампліфікації проведені за допомогою Fast Real-time PCR System (Applied Biosystems, США), у кінцевій реакції об’ємом 20 мкл, який містив 2X TaqMan універсальний Master Mix (Applied Biosystems, США), C___910351_10 rs510432 і матричну ДНК. Ампліфікація фрагментів генів складалася зі стадії денатурації при 95 °С протягом 20 с, а потім 40 циклів ампліфікації при 95 °С протягом 3 с і 60 °С протягом 30 с. Аналіз даних проводився з 7500 Fast Real-Time PCR Software.
Статистичний аналіз
Отримані результати були опрацьовані програмою SPSS (версії 22.0) та програмного середовища R (версії 3.0). Під час дослідження застосовано веб-програмне забезпечення SNP Analyzer (http://snpanalyzer.uthsc.edu). Відхилення від закону Харді — Вайнберга та асоціацію розвитку атопічних захворювань за наявності однонуклеотидного поліморфізму гена ATG5 досліджувалося за допомогою тесту χ2. Для вибору моделі, згідно з якою успадковується фенотипова ознака АЗ, використаний критерій Айкайке. Вірогідність розвитку АЗ при патологічних варіантах поліморфізму гена ATG5 визначалась за методом логістичної регресії. Для опрацювання даних, отриманих при обстеженні хворих дітей, використаний статистичний пакет Medstat.
Результати
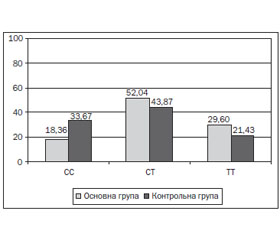
Під час дослідження отримані наступні результати: у 33 дітей (33,67 %) контрольної групи виявлена мажорна гомозигота СС, у 43 (43,87 %) — гетерозиготний варіант СТ, у 21 (21,43 %) — мінорний варіант ТТ поліморфізму rs510432 гена ATG5. У 18 дітей (18,36 %) основної групи була наявна мажорна гомозигота СС, у 51 (52,04 %) — гетерозиготний варіант СТ, у 29 (29,60 %) — мінорний варіант ТТ поліморфізму rs510432 гена ATG5 (рис. 1). За допомогою χ2-тесту знайдено статистично значущі відмінності у розподілі генотипів в основній і контрольній групах (χ2 = 6,36; р < 0,05).
Вивчена вірогідність розвитку атопічних захворювань у дітей залежно від поліморфізму rs510432 гена ATG5 з метою можливості використання даного генетичного тесту для ранньої діагностики, проведення профілактичних заходів та своєчасного призначення лікування.
Для розрахунку ризику розвитку АЗ у дітей використано формулу:
де е — число Ейлера (основа натуральних логарифмів);
де Bo — константа, В — коефіцієнт регресії при кожному алельному варіанті, Х — незалежна змінна (генотип).
Незалежні змінні кодувалися наступним чином: Х1 — генотип СС, Х2 — генотип СТ, Х3 — генотип ТТ. Результати аналізу наведені у табл. 1.
Підставивши у формулу результати, отримані за методом логістичної регресії, отримуємо:
z = –0,658174 + 0,875469 × Х1 + –0,742473 × Х2 + –2,360415× Х3.
Таким чином, отримана математична модель для обчислення ймовірності розвитку АЗ залежно від обраних нами факторів. Результати наведено в табл. 2.
Отже, за результатами логістичної регресії, ризик розвитку АЗ у дітей з генотипом СС rs510432 гена ATG5 становить 29,41 %, у гетерозигот СТ — 67,92 %, у дітей з мінорним генотипом ТТ досягає 72,04 %.
В основній групі 25 хворих (25,51 %) мали тяжку персистуючу бронхіальну астму (1-ша підгрупа), інші 73 дитини (74,49 %) мали інтермітуючу, легку та середньотяжку бронхіальну астму (2-га підгрупа). Розподіл алельних варіантів rs510432 гена ATG5 залежно від тяжкості захворювання наведено в табл. 3.
Для дослідження впливу генотипу поліморфізму rs510432 гена ATG5 на показники об’єму форсованого видиху за 1 секунду (ОФВ1) основна група була поділена на три підгрупи. До 1-ї підгрупи увійшло 18 хворих (18,37 %) із мажорним генотипом, до 2-ї — 51 дитина (52,04 %) із гетерозиготним генотипом, до 3-ї — 29 осіб (29,59 %) із мінорним генотипом. Дані щодо середніх значень показника ОФВ1 у підгрупах наведені у табл. 4.
Статистично значущих відмінностей між середніми значеннями показників ОФВ1 у пацієнтів із мажорним і гетерозиготним генотипом, гетерозиготним і мінорним генотипом не виявлено (р < 0,05). Середні значення ОФВ1 у хворих із мінорним генотипом вірогідно нижчі, ніж з мажорним генотипом (Q = 2,71; р < 0,05).
Для виявлення впливу зазначеного поліморфізму на ступінь тяжкості атопічного дерматиту серед дітей основної групи ми виділили підгрупу з 65 хворих (66,33 %) із легким ступенем тяжкості дерматиту та в стадії ремісії. Інші 33 дитини (33,67 %) з атопічним дерматитом середньої та тяжкої тяжкості становили 2-гу підгрупу. Розподіл алельних варіантів в обох підгрупах наведено в табл. 5.
Наступним етапом дослідження було з’ясування, чи має поліморфізм rs510432 гена ATG5 вплив на тяжкість алергічного риніту у дітей. Від даної патології страждала 31 дитина з основної групи, із них у 9 хворих (9,18 %) встановлено середньотяжкий/тяжкий алергічний риніт, у 22 дітей (22,45 %) — легкий ступінь тяжкості. Відповідно ці діти становили 1-шу та 2-гу підгрупу. Ми дослідили частоту алельних варіантів rs510432 гена ATG5 в обох підгрупах (табл. 6).
Обговорення та висновки
При порівнянні частоти алельних варіантів rs510432 гена ATG5 в українській, європейській популяціях та серед населення Азії і Африки виявлено, що розподіл алелей зазначеного поліморфізму відповідає європейському. Так, мажорний генотип у європейській популяції зустрічається у 31,86 % населення, гетерозиготний — у 44,24 %, патологічні гомозиготи — у 23,89 %. В азіатів мажорні гомозиготи СС виявляються у 12,79 % населення, гетерозиготи СТ — у 61,62 %, мінорний варіант ТТ — у 25,58 %. В афроамериканців зазначені алельні варіанти rs510432 гена ATG5 виявляються у 2,65, 27,43 та 69,91 % відповідно (NCBI dbSNP, Short Genetic Variations, https://ncbi.nlm.nih.gov/projects/SNP_ref.cgi?rs=510432). Наведені дані свідчать про неоднорідність розподілу алельних варіантів rs510432 гена ATG5 залежно від раси та унеможливлюють екстраполяцію результатів щодо впливу поліморфізму rs510432 гена ATG5 на розвиток атопічних захворювань в інших популяціях.
Встановлено, що мінорна алель Т rs510432 гена ATG5 асоціюється з підвищеним ризиком розвитку атопічних захворювань у дітей української популяції (χ2 = 6,36; р < 0,05). За результатами логістичної регресії ризик розвитку атопічних захворювань у дітей з мінорним генотипом досягає 72 %, що у 2,4 раза вище, ніж у дітей із мажорним генотипом. На нашу думку, отримані результати дають підстави рекомендувати використання даного генетичного тесту для дітей з випадками атопії в сімейному анамнезі для проведення профілактичних заходів і запобігання маніфестації «атопічного маршу».
Згідно з літературними даними, зміни автофагічної активності призводять до ремоделювання дихальних шляхів внаслідок структурних змін війчастого епітелію та активації транформуючого фактора росту β1 [23]. Отримані результати свідчать, що мінорний генотип поліморфізму rs510432 гена ATG5 імовірно сприяє ремоделюванню дихальних шляхів у дітей, хворих на бронхіальну астму, що призводить до зниження рівня ОФВ1.
Статистично значущих відмінностей у розподілі генотипів rs510432 гена ATG5 залежно від ступеня тяжкості атопічних захворювань не виявлено. Тяжкість перебігу даної групи захворювань обумовлена не лише генетичними факторами, але й більшою мірою впливом навколишнього середовища, адекватністю і тривалістю терапії до моменту включення у дослідження тощо.
Таким чином, поліморфізм rs510432 гена ATG5 може бути використаний для прогнозування ризиків розвитку атопічного маршу у дітей для проведення своєчасних профілактичних заходів. Окрім того, використання поліморфізму rs510432 гена ATG5 як предиктора зниження показників функції зовнішнього дихання у дітей дозволить індивідуально призначати лікування з метою запобігання розвитку бронхіальної астми. З іншого боку, застосування даного генетичного тесту для прогнозування тяжкості атопічних захворювань є недоцільним. Подальші дослідження є необхідними для з’ясування впливу поліморфізму на ефективність терапії атопічних захворювань.
Висновок
У дітей із мінорним генотипом ТТ поліморфізму гена ATG5 виявлено підвищений ризик розвитку атопічних захворювань, що становить 72 %. У хворих на бронхіальну астму мінорний генотип ТТ поліморфізму гена ATG5 асоційований зі зниженим показником ОФВ1. Поліморфізм rs510432 гена ATG5 доцільно використовувати з метою прогнозування розвитку атопічних захворювань у дітей.
Конфлікт інтересів. Автор заявляє про відсутність конфлікту інтересів.
Список литературы
1. Ласиця О.Л. Алергологія дитячого віку / О.Л. Ласиця, Т.С. Ласиця, С.М. Недельська. — К.: Книга плюс, 2004. — 368 с.
2. Охотникова Е.Н. Механизмы формирования и клинические особенности течения «атопического марша» у детей / Е.Н. Охотникова // Здоровье Украины. Тематический выпуск. — 2010. — С. 16-17.
3. Рекен М. Наглядная аллергология: Пер. с англ. / М. Рекен, Г. Греверс, В. Бургдорф. — М.: БИНОМ. Лаборатория знаний, 2013. — 238 с.
4. Beck L.A. Allergen sensitization through the skin induces systemic allergic responses / Beck L.A., Leung D.Y. // J. Allergy Clin. Immunol. — 2000. — № 106(5 Suppl.). — P. 258-63.
5. Betz C. Where is mTOR and what is it doing there? / Betz C., Hall M.N. // J. Cell. Biol. — 2010. — Vol. 203, № 4. — P. 563-574.
6. Cookson W.O. Genetic linkage of childhood atopic dermatitis to psoriasis susceptibility loci / Cookson W.O., Ubhi B., Lawrence R., Abecasis G.R., Walley A.J., Cox H.E., Coleman R., Leaves N.I., Trembath R.C., Moffatt M.F., Harper J.I. // Nat. Genet. — 2001. — Vol. 27, № 4. — P. 372-3.
7. Deretic V. Autophagy in infection / Deretic V. // Curr. Opin. Cell. Biol. — 2010. — Vol. 22, № 2. — P. 252-562.
8. Eichenfield L.F. Atopic dermatitis and asthma: parallels in the evolution of treatment / Eichenfield L.F., Hanifin J.M., Beck L.A., Lemanske R.F. Jr, Sampson H.A., Weiss S.T., Leung D.Y. // Pediatrics. — 2003. — Vol. 111, № 3. — P. 608-16.
9. Gupta J. Resolving the etiology of atopic disorders by using genetic analysis of racial ancestry / Gupta J., Johansson E., Bernstein J.A., Chakraborty R., Khurana Hershey G.K., Rothenberg M.E., Mersha T.B. // J. Allergy Clin. Immunol. — 2016. — pii: S0091-6749(16)30256-1. — doi: 10.1016/j.jaci.2016.02.045. [Epub ahead of print].
10. Hussey S. Autophagy as an emerging dimension to adaptive and innate immunity / Hussey S., Travassos L.H., Jones N.L. // Semin. Immunol. — 2009. — № 21 — P. 233-41.
11. Inoue D. Inducible disruption of autophagy in the lung causes airway hyper-responsiveness / Inoue D., Kubo H., Taguchi K., Suzuki T., Komatsu M. // Biochem. Biophys. Res. Commun. — 2011. — № 405. — P. 13-18.
12. Jyothula S.K. Autophagy and role in asthma / Jyothula S.K., Eissa N.T. // Curr. Opin. Pulm. Med. — 2013. — № 19. — P. 30-35.
13. Kondo N. Pharmacogenetics of asthma in children / Kondo N., Matsui E., Nishimura A., Kaneko H. // Allergy Asthma Immunol. Res. — 2010. — Vol. 2, № 1. — P. 14-9.
14. Lee J.U. Gene-Environment Interactions in Asthma: Genetic and Epigenetic Effects / Lee J.U., Kim J.D., Park C.S. // Yonsei Med. J. — 2015. — Vol. 56, № 4. — P. 877-86.
15. Levine B. Unveiling the roles of autophagy in innate and adaptive immunity / Levine B., Deretic V. // Nat. Rev. Immunol. — 2007. — Vol. 7, № 10. — P. 767-77.
16. Li G. Ménage à Trois in stress: DAMPs, redox and autophagy / Li G., Tang D., Lotze M.T. // Semin. Cancer Biol. — 2013. — Vol. 23, № 5. — P. 380-90.
17. Li C. Autophagy is induced in CD4+ T cells and important for the growth factor-withdrawal cell death / Li C., Capan E., Zhao Y., Zhao J., Stolz D., Watkins S.C., Jin S., Lu B. // J. Immunol. — 2006. — № 177. — P. 5163-5168.
18. Miller B.C. The autophagy gene ATG5 plays an essential role in B lymphocyte development / Miller B.C., Zhao Z., Stephenson L.M., Cadwell K., Pua H.H., Lee H.K., Mizushima N.N., Iwasaki A., He Y.W., Swat W., Virgin H.W. // Autophagy. — 2008. — Vol. 4, № 3. — P. 309-14.
19. Mizushima N. Autophagosome formation in mammalian cells / Mizushima N., Ohsumi Y., Yoshimori T. // Cell. Struct. Funct. — 2002. — Vol. 27, № 6. — P. 421-9.
20. Nakano H. An unexpected role for autophagy in degranulation of mast cells / Nakano H., Ushio H. // Autophagy. — 2011. — Vol. 7, № 6. — P. 657-9.
21. Oh J.Е. Autophagy as an innate immune modulator / Oh J.Е., Lee H.K. // Immune Network. — 2013. — Vol. 13, № 1. — P. 1-9.
22. Ortiz R.A. Genetics of allergic diseases / Ortiz R.A., Barnes K.C. // Immunol. Allergy Clin. North Am. — 2015. — Vol. 35, № 1. — P. 19-44.
23. Poon A. ATG5, autophagy and lung function in asthma / Poon A., Eidelman D., Laprise C., Hamid Q. // Autophagy. — 2012. — Vol. 8, № 4. — P. 694-5.
24. Puleston D.J. Autophagy in the immune system / Puleston D.J., Simon A.K. // Immunology. — 2014. — Vol. 141, № 1. — P. 1-8.
25. Schmid D. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes / Schmid D., Pypaert M., Munz C. // Immunity. — 2007. — № 26. — P. 79-92.
26. Spergel J.M. Atopic dermatitis and the atopic march / Spergel J.M., Paller A.S. // J. Allergy Clin. Immunol. — 2003. — № 112 (6 Suppl.). — P. 118-27.
27. Tao Zheng. The Atopic March: Progression from Atopic Dermatitis to Allergic Rhinitis and Asthma / Tao Zheng, Jinho Yu, Min Hee Oh, Zhou Zhu // Allergy Asthma Immunol. Res. — 2011. — Vol. 3, № 2. — P. 67-73.
28. Valdor R. Autophagy and the regulation of the immune response / Valdor R., Macian F. // Pharmacological. Research. — 2012. — № 66. — P. 475-483.
29. Xu Yi Autophagy in Innate and Adaptive Immunity / Xu Yi Eissa N.T. // Proc. Am. Thorac. Soc. — 2010. — Vol. 7. — P. 22-28.
30. Zhifen Yang. An Overview of the Molecular Mechanism of Autophagy / Zhifen Yang, Klionsky D.J. // Curr. Top. Microbiol. Immunol. — 2009. — № 335. — P. 1-32.

/16.jpg)
/17.jpg)
/17_2.jpg)
/17_3.jpg)
/17_4.jpg)
/18.jpg)