Гіперурикемія (ГУ) — підвищення рівня сечової кислоти (СК) у сироватці крові понад 360 мкмоль/л (6 мг/дл) у жінок и понад 420 мкмоль/л (6,8 мг/дл) у чоловіків. При цьому рівні можна спостерігати кристалізацію урату мононатрію у водному розчині при фізіологічному рівні рН та температурі тіла 36–36,6 °С [1]. Розчинність сечової кислоти у воді та фізіологічних рідинах обмежена і залежить переважно від рН та температури. Розчинність у воді при концентрації 6,8 мг/дл (404 мкмоль/л) відбувається при температурі 37 °С. Проте розчинність значно зменшується при низьких температурах і підкисленні, так, при 35 °С розчинність відбувається при концентрації СК 6,0 мг/дл (360 мкмоль/л), а при 30 °С — лише приблизно 4 мг/дл (238 мкмоль/л) [2]. З огляду на це кристали урату мононатрію спочатку потрапляють у тканини, що менш підігріваються та погано васкуляризуються (сухожилля, зв’язки) або не васкуляризовані (хрящ) [1]. Іn vivo утворення та осадження кристалів урату залежить не тільки від концентрації СК, рН і температури, але й регулюється концентрацією хлориду натрію та наявністю протеогліканів.
Гіперурикемія може бути пов’язана зі збільшенням продукції пуринів (наприклад, викликаних інтенсивним лізисом клітин), надмірним вживанням і абсорбцією пуринів, порушенням екскреції сечової кислоти. Близько двох третин від загальної кількості уратів у організмі залежить від ендогенної продукції (мієлопроліферативні та лімфопроліферативні захворювання, поліцитемія, пухлини, гемолітична анемія та під час хіміо- та радіотерапії) та недостатньої елімінації сечової кислоти, тоді як решта залежить від дотримання дієти, богатої на пурини [3]. Близько 70 % уратів елімінується нирками, решта — шлунково-кишковим трактом [4].
Безсимптомна гіперурикемія характеризується як стан підвищеної концентрації сечової кислоти (> 6,8 мг % (404 мкмоль/л)) без будь-яких симптомів подагри [5]. Безсимптомне підвищення СК у сироватці крові зустрічається у 5–8 % населення в популяції, при цьому тільки у 5–20 % з них розвивається подагра [6].
Дисфункція ендотелію та системне запалення є основними механізмами, які впливають на формування ефектів гіперурикемії [7, 8]. Запальний процес має системний характер і ініціюється кристалами урату, що елімінуються фагоцитами, нейтрофілами та макрофагами, крім того розвитку запалення сприяють вади вродженої системи імунітету. Мікрокристали покриваються опсонізуючими білками, що призводить до активації 2-го і 4-го toll-like-рецепторів і ініціації запального процесу. Прозапальні цитокіни, що виділяються моноцитами та синовіоцитами, трансформують локальний процес у системне запалення [9]. Вважається, що хронічне системне запалення викликає ушкодження ендотелію кровоносних судин. Внаслідок цього синтез оксиду азоту припиняється у стінці судини й активізується ренин-ангіотензин-альдостеронова система (РААС), що призводить до гіпертрофії м’язової оболонки судин (включаючи проліферацію та міграцію міоцитів) [10]. Крім того, хронічний системний запальний процес ініціює склеротичні зміни в судинах і тканинах [11].
У наш час такий патологічний стан, як гіпер-урикемія, викликає пристальну інтерес дослідників, що обумовлено асоціацією підвищеного рівня СК з розвитком низки патологій, які належать до так званих хвороб цивілізації: ожиріння, гіпертонічна хвороба, цукровий діабет (ЦД). Крім того, гіперурикемія є незалежним фактором розвитку серцево-судинних захворювань і корелює з різними ураженнями нирок.
Нещодавні епідеміологічні дослідження доводять, що проблема гіперурикемії стосується багатьох мільйонів людей (рис. 1).
Зростаюча частота гіперурикемії, швидше за все, спричинена вестернізацією способу життя та станом навколишнього середовища. У найближчому майбутньому через ожиріння та метаболічні розлади, а також старіння людського населення слід очікувати зростання частоти гіперурикемії [12, 13]. Це явище пов’язане зі стрімким економічним розвитком і зміною способу життя суспільства з вищим соціально-економічним статусом [14]. Підвищена поширеність гіперурикемії обумовлена такими загальноприйнятими звичками, як масивне споживання багатих пурином продуктів (м’яса, поживних речовин, морепродуктів), фруктози, зловживання алкоголем, прийом невеликих доз аспірину та тіазидів. Метаболічні розлади є фактором ризику гіпер-урикемії, близько 50 % пацієнтів з гіперурикемією мають симптоми метаболічного синдрому. Вона тісно пов’язана з діабетом, ожирінням, коронарним захворюванням, артеріальною гіпертензією (АГ) [3, 15, 16].
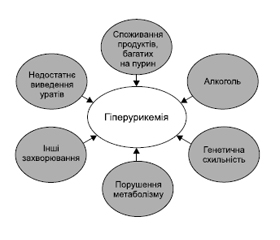
Схема основних факторів ризику гіперурикемії подана на рис. 2.
Давно відомо, що антиоксидантні властивості сечової кислоти можуть запобігти старінню, окиснювальному стресу та ушкодженню клітин [8, 18]. Проте останні клінічні та епідеміологічні дані свідчать про негативний вплив підвищеної концентрації сечової кислоти у сироватці крові [19]. Гіперурикемія відіграє важливу патофізіологічну роль у розвитку АГ, ЦД 2-го типу та є незалежним серцево-судинним фактором ризику [3, 5, 20], супроводжує метаболічний синдром, дисліпідемію, хронічне ниркове захворювання, ожиріння, рівень СК у сироватці суттєво змінюється залежно від статі та попереднього застосування діуретиків.
Найпоширеніші серцево-судинні захворювання та фактори, пов’язані з підвищеною концентрацією сечової кислоти, подано нижче.
Хвороби серцево-судинної системи та фактори, пов’язані з підвищеною концентрацією сечової кислоти [17]:
— Артеріальна гіпертензія та прегіпертензія.
— Захворювання нирок (мікроальбумінурія, знижена швидкість клубочкової фільтрації (ШКФ).
— Метаболічний синдром.
— Синдром обструктивного нічного апное.
— Захворювання периферійних судин.
— Інсульт, транзиторні порушення кровообігу.
— Прееклампсія.
— Підвищення рівня гострофазних реагентів (С-реактивний білок, активатор плазміногену тканин та ін.).
— Дисфункція ендотелію.
— Оксидативний стрес.
— Похилий вік.
— Чоловіча стать.
У жінок гіперурикемія зустрічається в постменопаузі (естрогени сприяють нирковій екскреції сечової кислоти [21], і ризик ускладнень збільшується при ще більш низькому рівні сечової кислоти в сироватці крові порівняно з чоловіками та потребує більшої уваги [22].
Перші публікації, присвячені вивченню взаємодії та зв’язку АГ і ГУ, з’явилися близько півтора століття тому. Ще в 70-ті роки ХІХ ст. F.A. Mahomed [23] у статті, присвяченій опису синдрому есенціальної гіпертензії, зауважив, що чимало пацієнтів з АГ народилися у сім’ях, члени яких страждали від подагри, і припустив, що причиною розвитку АГ є СК. Через десять років після цього ця гіпотеза знайшла підтвердження в дослідженні А. Haig [24], який застосовував дієту з низьким умістом пуринів для запобігання розвитку АГ і серцево-судинних захворювань (ССЗ). У цей же час французький академік H. Huchard [25] при гістологічному дослідженні в осіб, які страждали від АГ, виявив нирковий артеріолосклероз, що спостерігався переважно у трьох групах хворих: при подагрі, при надмірному вживанні жирного м’яса і при свинцевому отруєнні — все це мало зв’язок з ГУ.
Ці припущення знайшли підтвердження і в наш час. За даними досліджень, проведених у 60-ті та 70-ті роки ХХ ст., ГУ мала місце приблизно у 5 % населення США [26], а у хворих на АГ підвищений рівень СК виявлено у 40–60 % випадків, також АГ визначалася у 50–65 % хворих на подагру [27].
Дані дослідження Bogalusa Heart Study (дослідження серцево-судинних факторів ризику (ФР), що виявляються в дитинстві) дали можливість вивчити передбачуваність розвитку АГ у дорослих залежно від рівня СК у дитячому віці [28]. Зроблено припущення, що раннє підвищення рівня CК може відіграти ключову роль у розвитку АГ. Дослідження включало 577 осіб (серед них 333 жінки) віком від 5 до 17 років. Середній період спостереження становив 11,4 року (діапазон — від 7,4 до 15 років). У 23 дітей у подальшому розвинулась АГ. В дитинстві у них був більш високий рівень СК порівняно з 554 обстеженими з нормальним рівнем артеріального тиску (АТ) (5,12 мг/дл проти 4,30 мг/дл; p = 0,007). Виявлено значущий зв’язок між рівнем СК і значеннями систолічного (САТ) (r = 0,31; p ≤ 0,0001) і діастолічного (ДАТ) АТ (r = 0,20; p ≤ 0,0001) в дитинстві. Крім того, виявлено кореляцію між базальним рівнем СК і показниками САТ (r = 0,29; p ≤ 0,0001) і ДАТ (r = 0,28; p ≤ 0,0001) у дорослих у цілому по групі з урахуванням поправки на вік, стать, расову приналежність, індекс маси тіла (ІМТ) у дитинстві [28].
Більше того, у пацієнтів з АГ, асоційованою з метаболічним синдромом, спостерігається позитивна кореляція між концентрацією сечової кислоти та ІМТ, концентрацією інсуліну натщесерце та показником інсулінорезистентності HOMA (IR) [3]. Гіперурикемія часто зустрічається у пацієнтів із пороговими значеннями АТ, особливо якщо вона асоційована з протеїнурією [15, 16].
Lee та співавт. [29] показали, що пацієнти з прегіпертензією (САТ 120–140 мм рт.ст. або ДАТ 80–90 мм рт.ст.) і мікроальбумінурією мали більш високі концентрації сечової кислоти порівняно з людьми з нормальною альбумінурією (чоловіки — від 6,5 ± ± 1,1 мг/дл (387 ± 65 мкмоль/л) і до 6,2 ± 1,1 мг/дл (369 ± 65 мкмоль/л) відповідно; р = 0,017; жінки — 4,8 ± 0,9 мг/дл (286 ± 54 мкмоль/л) проти 4,4 ± ± 0,9 мг/дл (262 ± 54 мкмоль/л); р = 0,006).
Результати дослідження дають можливість припустити, що рівень СК робить істотний внесок у серцево-судинний ризик у пацієнтів з нелікованною АГ. Продемонстровано, що ГУ підвищує ризик виникнення та прогресування АГ [30]. Подібні дані були отримані Y. Taniguchi і співавт. [31]. В аналіз було включено 6356 жителів Японії чоловічої статі віком 35–60 років із САД < 140 мм рт.ст. і ДАТ < 90 мм рт.ст. За період 61 716 людино-років було зареєстровано 639 випадків розвитку АГ. Після виключення традиційних ФР розвитку ССЗ рівень CК виявився тісно пов’язаним з підвищеним ризиком розвитку АГ.
Також результати дослідженнь дозволяють припустити, що рівень СК робить істотний внесок у серцево-судинний ризик у пацієнтів з нелікованою АГ. P. Grayson і співавтори [32] провели метааналіз, який включав 18 когортних досліджень за участю 55 607 осіб без АГ на початку досліджень. Період спостереження становив від 3 до 21 року. Виявлено, що ГУ асоціюється з підвищеним ризиком АГ (відносний ризик (ВР) 1,41; 95% довірчий інтервал (ДІ) від 1,23 до 1,58). Причому збільшення рівня СК на 1 мг/дл супроводжувалося збільшенням ризику розвитку АГ на 13 %. Відмічено значне збільшення ризику у молодих людей (р = 0,02) і у жінок (р = 0,059).
Викликають інтерес результати Фрамінгемського дослідження [33] (3329 осіб, серед них жінок — 55,6 %, середній вік — 48,7 року), які продемонстрували, що підвищення рівня СК є предиктором розвитку АГ. З дослідження виключалися пацієнти, які страждали від АГ, які перенесли ІМ, а також мали серцеву або ниркову недостатність, подагру. Внаслідок цього за 4 роки після початку дослідження АГ розвинулася у 458 (13,8 %) обстежених, підвищення АТ (порівняно з початковим) зазначалося у 1201 (36,1 %) хворого. При багатофакторному аналізі з поправкою на вік, стать, ІМТ, наявність ЦД, куріння, вживання алкоголю, рівень креатиніну, протеїнурію, ШКФ підвищення рівня СК асоціювалося з ризиком розвитку АГ (відношення шансів (ВШ) — 1,17; 95% ДІ 1,02–1,33) і збільшенням АТ порівняно з початковим (ВШ — 1,11; 95% ДІ 1,01–1,23).
Є докази, що у популяції пацієнтів з дуже високим ризиком ССЗ концентрація СК у сироватці крові є незалежним показником смертності. У дослідженні, опублікованому Ndrepepa і співавт. [34] група з 5124 пацієнтів із гострими коронарними синдромами (1629 інфарктів серця з підйомом сегмента ST, 1332 без підйому сегмента ST і 2163 з нестабільною коронарною хворобою) була розподілена на групи відповідно до концентрації СК у сироватці крові: 1-ша група — 1,3–5,3 мг/дл (77–315 мкмоль/л); 2-га — 5,3–6,3 мг/дл (315–375 мкмоль/л); 3-тя група — 6,2–7,5 мг/дл (375–446 мкмоль/л); 4-та група — від 7,5–18,4 мг/дл (446–1094 мкмоль /л). Через рік спостереження було зареєстровано 80 смертей у 1-й групі, 77 — у 2-й, 72 — у 3-й та 122 — у 4-й групі. Ризик смертності, що не коригується, становив 3,05 (95% ДІ 2,54–3,67; р < 0,001) для 4-ї групи порівняно з 1-ю. Варто зазначити, що кореляція між концентрацією сечової кислоти та смертністю залишається значною (збільшення на 12 % ризику смертності на рік для 1 мг/дл (59 мкмоль/л) сечової кислоти).
Ще одне дослідження доводить, що ГУ робить значний внесок у збільшення ризику серцево-судинних катастроф. Зокрема, в дослідженні NHANES I (National Health Аnd Nutrition Examination Survey) показано зв’язок між ГУ та зростанням серцево-судинної летальності [35]. З підвищенням рівня СК ризик смерті від ішемічної хвороби серця (ІХС) зростав на 77 % у чоловіків і на 300 % у жінок. Збільшення концентрації СК на 1 мг/дл (59,5 мкмоль/л) асоціюється зі значним підвищенням летальності від ССЗ як серед чоловіків, так і серед жінок [35]. У дослідженні PIUMA [36], в яке було включено понад 1500 пацієнтів із нелікованою раніше АГ, також продемонстровано, що рівень СК сироватки є важливим предиктором розвитку ССЗ і смертності від них.
Слід зазаначити, що, незважаючи на численні дані про зв’язок ГУ з ССЗ, саме наявність подагри є незалежним ФР інфаркту міокарда (ІМ) [2]. Подагра — системне тофусне захворювання, що характеризується відкладенням у різних тканинах кристалів моноурату натрію, розвитком через це запалення у хворих на ГУ, обумовлену зовнішньосередовищними та/або генетичними факторами.
Тенденцією сьогодення є більш агресивний клінічний перебіг подагри, що проявляється великою кількістю залучених суглобів, наявністю нефролітіазу, частим переходом у хронічний артрит. Зросла частота жіночої та сімейної подагри, нефролітіазу [37, 38].
Провідною причиною смерті хворих на подагру є ССЗ, обумовлені атеросклеротичним ураженням судин [39]. Накопичено численні дані про високу поширеність коморбідних патології серед хворих на подагричний артрит, зокрема, показано високу захворюваність на АГ, ІХС, інсульт, атеросклеротичне ураження сонних артерій, судинну деменцію [40].
Результати дослідження MRFIT [41] свідчать про підвищений ризик розвитку ІМ у пацієнтів із подагрою після виключення класичних ФР. Можна припустити, що це — наслідок хронічного запалення, що є у цих хворих. В одному 12-річному дослідженні [42] взаємозв’язку АГ, ожиріння, використання діуретиків і частоти виникнення подагри (47 150 чоловіків) при багатофакторному аналізі виявлена асоціація між АГ (ВШ 2,31; 95% ДІ 1,96–2,72), прийомом діуретиків (ВШ 1,77; 95% ДІ 1,42–2,20) і ризиком розвитку подагри. Після виключення з аналізу осіб, які вживали діуретики, зв’язок між рівнями СК і АГ істотно не змінився (ВШ 2,28; 95% ДІ 1,93–2,70).
Патогенетичний зв’язок ГУ і АГ продемонстровано в низці експериментальних робіт на тваринах. Так, R.J. Johnson і співавт. [43, 44] показали in vivo, що помірне підвищення рівня СК може призводити до гломерулотубулярних ушкоджень, сприяє активізації ренин-ангіотензин-альдостеронової системи (РААС) і підвищенню АТ. При цьому всі зміни зазнавали зворотного розвитку після усунення ГУ. В експерименті на щурах Т. Nakagawa і співавт. [45] виявили чіткий зв’язок між м’якою ГУ і гіпертрофією ниркових клубочків через активізацію РААС. При збільшенні рівня СК на 1 мг/дл САТ підвищувався на 30 мм рт.ст., розвивалася гіпертрофія гломерулярного клубочка.
Встановлено, що внаслідок надмірної продукції СК нирки компенсаторно збільшують виведення уратів із сечею, що може призводити до розвитку різних нефропатій, зокрема уратного тубулоінтерстиційного нефриту, який рідко діагностується через мінімальні прояви [46]. Продемонстровано, що розвиток ГУ асоціюється зі станом ниркової гемодинаміки, причому вважають, що її погіршення передує розвитку порушення метаболізму СК і нефропатії.
ГУ також асоціюється з рядом патологічних станів — ендотеліальною дисфункцією, уповільненням окислювального метаболізму, адгезією тромбоцитів, порушенням реології крові та агрегацією. M. Mazzali і співавт. [47] у дослідах in vivo довели, що ізольована ГУ призводить до виникнення АГ, зумовленої ендотеліальною дисфункцією й активізацією РААС, яка згодом трансформується в нефрогенну внаслідок розвитку артеріолосклеротичного ушкодження судин. Однак призначення на початку досліду інгібіторів ксантиноксидази (алопуринолу) або урикозуричних препаратів (бензбромарону) разом з оксонієвою кислотою сприяло нормалізації АТ і перешкоджало розвитку структурних змін у нирках [48]
У той же час, навпаки, АГ є ФР розвитку ГУ і, відповідно, подагри [49]. Внаслідок підвищення рівня АТ порушується стан мікроциркуляторного русла, що призводить до системної ішемії тканин, ураження клітин організму з масовим розпадом аденозинтрифосфату на аденін і ксантин та підвищення вироблення ксантиноксидази. Збільшення концентрації як субстрату (ксантіну), так і ферменту (ксантиноксидази) супроводжується надмірним утворенням СК [50]. Ще одним механізмом розвитку ГУ та подагри у хворих на есенціальну АГ є інтраренальна ішемія, що сприяє зниженню ниркового кровотоку, підвищенню рівня лактату в крові, який стимулює реабсорбцію, пригнічує секрецію уратів канальцевою аніонно-обмінної транспортною системою [51]. Інше можливе пояснення полягає в тому, що знижена екскреція СК може бути пов’язана з підвищеною реабсорбцією натрію і води в проксимальних ниркових канальцях, оскільки реабсорбція уратів, натрію і води здіснюється однією і тією ж транспортною системою [52].
Можна припустити, що АГ, призводячи до складних метаболічним зрушень, може бути як причиною ГУ і, відповідно, подагри, так і її наслідком. Сполучною ланкою між АГ і подагрою можуть служити й інші обмінні порушення, в тому числі компоненти метаболічного синдрому (МС). Відомо, що частота МС при подагрі набагато перевищує таку в популяції [53]. АГ — одна зі складових МС, і виникнення АГ патогенетично тісно пов’язано з іншими його симптомами (ІР, порушення вуглеводного обміну, ожиріння) [54].
У наш час ІР і гіперінсулінемія (ГІ) розглядаються як сполучна ланка між високим АТ і метаболічними порушеннями [55]. У хворих на АГ без ожиріння відзначається істотне зниження (на 30 %) чутливості до інсуліну, незважаючи на нормальну толерантність до глюкози [56]. У пацієнтів із підвищеним рівнем АТ ГІ виявлялася як натщесерце, так і при внутрішньовенному або пероральному навантаженні глюкозою [57]. Більше того, ГІ виявляється не тільки при вираженому підвищенні АТ, але і при високому нормальному рівні.
Більшість авторів [58] сходяться на думці, що існує кілька шляхів, які обумовлюють зв’язок АГ і ІР. Одним із механізмів, за допомогою якого інсулін може призводити до розвитку АГ, є здатність цього гормону активізувати симпатичну нервову систему [59]. За експериментальними даними, гіперкалорійне харчування, що стимулює розвиток ГІ, спричинює підвищення тонусу симпатичної нервової системи, а потім підвищення АТ як у тварин, так і в людей [60]. У осіб із початково нормальним рівнем АТ встановлено дозозалежну відповідність між приростом концентрації інсуліну і підвищенням вмісту норадреналіну в крові [60].
Тонус симпатичної нервової системи за наявності ГІ підвищується через активізацію не лише інсулінових, але й адренергічних рецепторів [61], що призводить до підвищення рівня катехоламінів у плазмі крові. Також ГІ сприяє активізації РААС, що викликає збільшення вироблення ангіотензину II, спазм гладких м’язів артеріол, підвищення гідростатичного тиску в клубочках, активізацію синтезу альдостерону, збільшення реабсорбції натрію, внаслідок чого збільшується об’єм циркулюючої крові, підвищується чутливість гладких клітин артеріол до впливу пресорних гормонів (ангіотезин II, катехоламіни) і розвивається АГ [62].
Необхідно звернути увагу ще на одну важливу причину розвитку АГ — абдомінально-висцеральне ожиріння, наявне у значної частини хворих на ГУ [63]. Результати епідеміологічних досліджень [42] свідчать про те, що ІМТ незалежно корелює з підвищеним АТ. Також доведено, що схуднення призводить до зниження АТ у хворих на ожиріння та сприятливо впливає на інші ФР (ІР, ЦД, гіперліпідемія, гіпертрофія лівого шлуночка (ГЛШ), обструктивне апное уві сні) [64].
У даний час висцеральну жирову тканину вважають самостійним ендокринних органом [65]. Адипоцити синтезують велику кількість учасників регуляції АТ і обмінних процесів біологично активних речовин. До них зараховують лептин, вільні жирні кислоти, інсуліноподібний фактор росту 1-го типу, ангіотензиноген, ангіотензин, інтерлейкіни, естрогени та ін. [66].
Вільні жирні кислоти, рівень яких натще в 2 рази вище в осіб з ожирінням, безпосередньо впливають на судини, викликаючи їх спазм, і в той же час — на центральні механізми, що регулюють активність симпатичної нервової системи [67]. Важливе місце в патогенезі АГ при ожирінні належить гіперлептинемії. Відзначено, що у хворих на ожиріння відбувається підвищення рівня лептину, який, за різними даними, може в 2–7 разів перевищувати норму [68]. Лептин має подібну до інсуліну дію на гіпоталамус і нирки, що призводить до збільшення активності симпатичної нервової системи і РААС [69]. Лептин чинить різноманітний вплив на клітини судин: індукує окислювальний стрес в ендотеліальних клітинах, підвищує продукцію моноцитарного хемоатрактного білка й ендотеліну-1 [70, 71], викликає міграцію, проліферацію і гіпертрофію тучних клітин [72], посилює кальцифікацію клітин судинної стінки і прискорює тромбоутворення, підвищуючи агрегацію тромбоцитів [73]. За даними епідеміологічних досліджень, лептин є ФР ІМ [74], геморагічного інсульту [75].
Відповідно, розвиток АГ у пацієнтів із надмірною масою тіла обумовлено як мінімум чотирма механізмами — активізацією симпатичної нервової системи, порушенням ендотеліальної функції, судинною вазоконстрикцією та підвищенням серцевого викиду. Отже, всі вищезазначені фактори можуть бути передумовою до розвитку АГ у хворих на ГУ та подагру з огляду на високу зустрічальність у них МС і його окремих компонентів.
Відомо, що підвищення АТ сприяє ураженню органів-мішеней, у тому числі ГЛШ, збільшенню товщини комплексу інтима-медіа сонних артерій, що є, в свою чергу, незалежним ФР серцево-судинних ускладнень і смертності [76]. У той же час висока частота у хворих на подагру додаткових ФР — ожиріння, ІР, ГУ, ЦД і МС — збільшує ризик розвитку ураження органів-мішеней. Так, у осіб з АГ та ожирінням таке ураження відбувається набагато раніше і більш виражено, ніж у хворих на АГ без ожиріння. Навіть у осіб без АГ ожиріння може призвести до зміни структурно-функціонального стану серця. При поєднанні ожиріння з АГ виникають додаткові зміни, що викликають перевантаження лівого шлуночка і розвиток його концентричної гіпертрофії. Для порівняння: частота ГЛШ у пацієнтів з АГ і нормальною масою тіла становить 28 %, без АГ, але з ожирінням — 22 %, а з АГ і ожирінням — 64 % [77].
В опублікованій роботі C.-F. Kuo і співавт. [78] продемонстрована асоціація ГУ зі збільшенням жорсткості судинної стінки (за значеннями кісточково-плечового індексу) і ГЛШ у чоловіків і жінок із поправкою на вік, протеїнурію, підвищений рівень С-реактивного білка, компоненти МС. Показано, що ГУ поряд з іншими ФР, пов’язаними з атеросклерозом, може відігравати роль у розвитку ГЛШ через збільшення жорсткості судинної стінки.
На думку багатьох дослідників, виникнення та прогресування ураження органів-мішеней обумовлене мікроциркуляторними порушеннями. Передбачається, що зміни мікросудин міокарда мають велике значення в розвитку ІХС, а також хронічної серцевої недостатності (ХСН) на тлі АГ і ГУ [79]. Невід’ємною частиною системи мікроциркуляції є клубочкові капіляри нирок. Порушення нейрогуморальної регуляції тонусу микросудин і зміни проникності базальної мембрани в процесі розвитку захворювання змінюються структурною перебудовою судин у нефроангіосклероз [79]. Ремоделювання мікроциркуляторного русла багато в чому обумовлює і поява церебральних ускладнень АГ: гіпертензивної енцефалопатії, гострого і минущого порушення мозкового кровообігу.
Було доведено, що підвищена концентрація сечової кислоти не тільки пов’язана з підвищеним ризиком захворювань серцево-судинної системи, але також сприяє розвитку когнітивних розладів у пацієнтів [80]. Результати епідеміологічного дослідження показують, що гіперурикемія може викликати деменцію. Ruggiero і співавт. [18] у крос-секційному дослідженні популяції 1016 осіб похилого віку довели, що при підвищеній концентрації сечової кислоти у сироватці крові існує високий ризик розвитку деменції.
Одним з основних місць, в яких спостерігаються антиоксидантні дії сечової кислоти, є центральна нервова система, особливо у випадку співіснування таких захворювань, як розсіяний склероз, хвороба Паркінсона або інсульт [81, 82]. Дослідження показують, що позитивна роль СК у запобіганні гострій активації оксидантами прозапальних клітин у крові може бути корисним механізмом, при якому концентрація СК викликає загальну антиоксидантну активність. З іншого боку, СК не може усунути всі оксигенні радикали, такі як супероксиди. Експерименти довели, що залежно від концентрації сечова кислота може перетворитися на прооксидант через активацію внутрішньоклітинного продукування супероксиду NADP оксидазою. Це так званий окисно-антиоксидантний парадокс [19]. СК через її деградацію та вищезгадані механізми може утворювати вільні радикали, що одночасно викликає запальну реакцію та дисфункцію ендотелію [83].
З огляду на високу поширеність АГ у хворих на подагру та ГУ, що є самостійним ФР серцево-судинних ускладнень і смертності, а також наявність додаткових ФР (надмірна маса тіла, дисліпідемія, ІР, ЦД), необхідно проведення ранньої діагностики АГ і виявлення ушкодження органів-мішеней (серця, нирок, судин) з метою профілактики розвитку серцево-судинних ускладнень. Вкрай важливо вчасно діагностувати АГ і призначати відповідне лікування, щоб зупинити розвиток ураження органів-мішеней і тяжких судинних ускладнень.
Добове моніторування АТ (ДМАТ) є одним з основних методів контролю АТ. ДМАТ суттєво полегшує діагностику АГ, дає можливість оцінити всі зміни АТ [84]. Доведено прогностичне значення у розвитку ураження органів-мішеней показників ДМАТ [85]: середніх значень САТ і ДАТ у денні та нічні години, пульсового АТ, індексів навантаження тиском, показників добового ритму АТ і варіабельності АТ, величини підвищення АТ у ранковий час.
Для діагностики ураження магістральних артеріальних судин при АГ проводять ультразвукову діагностику загальної сонної артерії, що допомагає виявити ознаки ремоделювання її стінки за збільшенням товщини комплексу інтима-медіа > 0,9 мм або наявності атеросклеротичної бляшки (товщина комплексу інтима-медіа > 1,3 мм) [86, 87]. Ехокардіографія є більш чутливим методом діагностики ГЛШ [88] та оцінки серцево-судинного ризику [89]. ГЛШ і збільшення товщини комплексу інтима-медіа сонних артерій є незалежним ФР серцево-судинних ускладнень і смертності [90]. Отже, ретельний моніторинг хворих на подагру та ГІ дозволяє своєчасно виявляти підвищення АТ і ураження органів-мішеней, проводити профілактіку розвитку ССЗ і кардіоваскулярних катастроф.
Актуальною проблемою є застосування патогенетично обґрунтованої, безпечної гіпотензивної терапії, що забезпечує адекватний контроль АТ протягом доби. У хворих на подагру, які страждають від АГ, необхідно брати до уваги взаємозв’язок складних метаболічних процесів, щоб не збільшити ризик терапевтично індукованої подагри [40]. Відомо, що деякі антигіпертензивні препарати можуть сприяти зростанню рівня CК у сироватці крові та, відповідно, підвищувати ризик розвитку подагри. Зростання вмісту СК у сироватці крові з розвитком суглобової подагри — добре відоме небажане явище, що виникає при застосуванні як тіазидних, так і петльових діуретиків [91]. Крім того, відзначено, що всі споріднені діуретики і спіронолактон також демонструють подібні ефекти [92].
Ситуація, що формується при стійкій ГУ, — кристалізація уратів у просвіті ниркових канальців, призводить до дисфункції нефрона та подальшого зниження діурезу, що створює враження резистентності до діуретиків. В епідеміологічному дослідженні [93] показано, що прийом петльових діуретиків обумовлює зростання ризику рецидивуючого подагричного артриту більше ніж у 3,5 раза. Виникнення ГУ, пов’язаної з прийомом петльових діуретиків, обумовлено тим, що ці препарати значною мірою зменшують нирковий кліренс сечової кислоти (СК) і її солей через розлад функції відповідних транспортних систем ниркових канальців [94]. Разом із тим доведено, що торасемід значно меншою мірою, ніж фуросемід і тіазидні діуретики, пригнічує функцію транспортера MRP-4, що локалізується на епітеліоцитах проксимальних канальців і бере участь в активній екскреції СК [95]. Отже, торасемід на відміну від інших петльових діуретиків та тіазидів меншою мірою пригнічує екскрецію СК і не настільки істотно сприяє наростанню урикемії.
У дослідженні [91], що включало аналіз 5789 пацієнтів з АГ, вивчено вплив різних доз діуретиків та їх комбінацій з іншими групами антигіпертензивних препаратів на рівень CК. Поширеність безсимптомної ГУ у пацієнтів з АГ у цьому дослідженні становила 21,6 %. Тривалість терапії в середньому була від 1,5 до 3 місяців. Встановлено, що стійке підвищення рівня CК у пацієнтів, які отримують комбіновану антигіпертензивну терапію, що включає гідрохлортіазид (ГХТ) у дозах 12,5 або 25 мг, спостерігається в 17,2 % випадків. У 13,3 % пацієнтів це підвищення визначається головним чином через недотримання гіпопуринової дієти та вживання алкоголю. У 3,9 % пацієнтів не вдалося виявити додаткових факторів підвищення рівня СК, крім початку антигіпертензивної терапії, що включає ГХТ у дозах 12,5 або 25 мг. Слід зазначити, що у 62,4 % пацієнтів спостерігалося транзиторне підвищення рівня CК через 2 тижні після початку комбінованої антигіпертензивної терапії, що включає ГХТ, більш виражене в групі жінок, із поверненням показників до вихідних через 3 місяці після закінчення терапії. При цьому не виявлено дозозалежного впливу ГХТ (12,5 або 25 мг), що входить до складу комбінованої терапії, на рівень CК. Вираженого впливу на рівень CК інших груп препаратів, що застосовуються для корекції АТ (інгібіторів АПФ, β-адреноблокаторів, антагоністів кальцію) в складі комбінованої антигіпертензивної терапії, що включає ГХТ у дозі 12,5 або 25 мг, не відзначено.
Крім відомих небажаних ефектів деяких діуретиків [42, 92], у короткострокових дослідженнях було показано гіперурикемічний ефект деяких β-блокаторів [92]. Так, пропранолол, атенолол і метопролол збільшують сироваткову концентрацію СК у хворих на АГ [92].
Продемонстровано, що блокатори кальцієвих каналів [96] і лозартан [97] знижують рівень СК, що може знижувати ризик розвитку подагри. Такі властивості блокаторів кальцієвих каналів можуть бути результатом їх впливу на функцію нирок. Показано збільшення екскреції СК під впливом блокаторів кальцієвих каналів [98]. Блокатори кальцієвих каналів можуть збільшувати ШКФ і, отже, швидкість виведення СК і креатиніну. Показано, що амлодипін збільшує вихід рідини з проксимальних канальців за допомогою суттєвого зменшення проксимальної канальцевої реабсорбції натрію і відповідного збільшення реабсорбції натрію в дистальних канальцях [99]. У дослідженні ACTION (A Coronary Disease Trial Investigating Outcome with Nifedipine GITS) [96] (n = 7665) встановлено, що ніфедипін, для якого характерна виражена судинорозширювальна дія на судини нирок, викликає зниження вмісту СК у сироватці крові та ризику подагри. Зазначені властивості амлодипіну і ніфедипіну можуть зменшувати ризик подагри на 21 і 13 % відповідно [42]. Передбачається, що аналогічного протективного ефекту можна очікувати і від дилтіазему. Встановлено, що у пацієнтів зі стенокардією, але без АГ, прийом блокаторів кальцієвих каналів також знижує ризик подагри на 16 % порівняно з особами, які приймали інші антиангінальні засоби [98]. Отже, терапія блокаторами кальцієвих каналів сприяла зниженню ризику подагри і у пацієнтів з АГ, і у осіб без підвищення АТ.
Інгібітори ангіотензинперетворюючого ферменту (АПФ) мають здатність збільшувати ниркову екскрецію СК через зниження реабсорбції в проксимальних канальцях [92]. Клінічно встановлено, що інгібітори АПФ — каптоприл [100], еналаприл [101], раміприл [92] і лізиноприл [92] — перешкоджають підвищенню рівня CК, спровокованого діуретиками. Однак такі ефекти мають не всі інгібітори АПФ. Так, встановлено підвищення рівня СК у сироватці крові на фоні терапії периндоприлом, особливо при збільшенні дози [102], що не перешкоджає розвитку гіперурикемії, асоційованої з прийомом діуретиків [102].
Аналіз дослідження Losartan Intervention for Reducing Endpoint (LIFE) виявив урикозуричні властивості лозартану. Також це дослідження продемонструвало, що вплив лозартану на рівень СК має позитивне клінічне значення: у хворих у групі лозартану частота серцево-судинних подій була менше, ніж у групі атенололу [103]. При багатофакторному аналізі даних результати були пояснені меншими змінами рівня СК у групі лозартану (від 328 до 348 ммоль/л) порівняно з групою атенололу, в якій рівень СК істотно підвищився (від 329 до 376 ммоль/л). Зниження рівня СК відбувається повільно, що запобігає можливому загостренню суглобового синдрому у хворих на подагру [104]. Це обумовлене двома механізмами лозартану: збільшенням тубулярної секреції (механізму, характерного для препаратів, що блокують ААС) та збільшенням канальцевої секреції СК (механізму, характерного для лозартану) [97]. Лозартан блокує дві основні транспортні системи епітеліоцитів дистальних канальців, що беруть участь у реабсорбції уратів (урат/лактат і урат/хлорид), і захищає структури ниркового тубулоінтерстицію від шкідливої дії уратів. Слід зазначити, що урикозурична дія лозартану не характерна для інших препаратів цього класу лікарських засобів. Так, у блокаторів ангіотензинових рецепторів, зокрема валсартану 80 мг на добу [105], кандесартану 8–16 мг на добу [106], телмісартану [107], ірбесартану [108], таких ефектів немає. Важливо підкреслити, що лозартан завдяки своєму гіпоурикемічному ефекту запропонований експертами Європейської ліги ревматологів для лікування хворих на АГ і подагру.
З практичної точки зору для контролю АТ при АГ у більшості пацієнтів необхідне призначення комбінації двох і більше препаратів, а у пацієнтів високого ризику лікування треба почати з комбінованої терапії. При цьому, якщо один препарат комбінації, що призначається, збільшує ризик подагри, а інший його зменшує, то в цілому ризик знижується. Однак якщо обидва препарати збільшують ризик, то в цьому випадку він подвоюється. Так, застосування блокаторів кальцієвих каналів (амлодипін, ніфедипін пролонгованої дії та ін.) у поєднанні з лозартаном значно знижує ризик подагри [109]. У разі необхідності призначення потрійної комбінованої терапії спостерігається подібний ефект. Відзначено, що в разі комбінованої терапії, яка включала діуретик та інгібітор АПФ, відносний ризик більш зростав при додаванні β-блокатора [42].
Лікарі повинні коригувати рівень СК у хворих із високим ризиком розвитку серцево-судинних подій навіть за наявності безсимптомної ГУ [110]. Алопуринол — препарат першої лінії в негіпотензивній терапії у пацієнтів з АГ та безсимптомною гіперурикемією. Алопуринол — це неселективний інгібітор ксантиноксидази, що пригнічує синтез сечової кислоти залежно від дози. Він вважається одним із найефективніших препаратів, що зменшують концентрацію сечової кислоти в сироватці крові [111]. Лікування слід починати з використання низької дози 100 мг на добу і збільшувати його на 100 мг кожні 2–4 тижні до максимальної дози 900 мг/до-бу при необхідності. Можна очікувати, що кожні 100 мг алопуринолу зменшать концентрацію СК на 1 мг/дл. Доза препарату завжди повинна бути корегована залежно від кліренсу креатиніну та приймоу інших ліків, які знижують концентрацію СК у сироватці крові [112]. Алопуринол зазвичай використовується не тільки завдяки своїй здатності зменшувати концентрацію СК, але також завдяки своєму доведеному захисному впливу на серцево-судинну систему. Вигідна роль терапії алопуринолом у зниженні смертності була підтверджена в дослідженні, проведеному у популяції пацієнтів із гіперурикемією [113]. У дослідженнях було доведено, що призначення алопуринолу було пов’язане зі зниженням загального рівня смертності [114, 115].
Алопуринол також викликав значне зниження САТ у пацієнтів, які приймали гіпотензивні препарати, а також у нелікованих. Зниження АТ може бути спричинено сприятливим впливом алопуринолу на жорсткість артерій і, як наслідок, зниження серцево-судинних ускладнень [116].
Інший препарат — фебуксостат, який є інгібітором ксантиноксидази і знижує концентрацію СК у сироватці, нещодавно був представлений в Україні [117]. Основним показанням до застосування фебуксостату є хронічна гіперурикемія, пов’язана із захворюваннями, які вже призвели до депозиції уратів (зокрема тофуси та активне запалення суглобів). Побічні ефекти фебуксостату, а також алопуринолу, зустрічаються рідко. Найтяжчі побічні ефекти — це випадки серйозного анафілактичного стану.
Отже, для хворих на ГІ та подагру характерна висока поширеність АГ, а проблема вибору антигіпертензивного препарату являє великі складності. При проведенні диференційованої антигіпертензивної терапії необхідно брати до уваги вплив на показники ліпідного та вуглеводного обміну, рівень СК, при цьому препарат повинен мати високу антигіпертензивну ефективнісь. При виборі терапії слід зважати на взаємозв’язок складних метаболічних процесів, щоб не збільшити ризик терапевтично індукованої подагри. Діуретики широко призначаються для лікування АГ, але вони є фактором ризику гіперурикемії і подагри. У пацієнтів з подагрою або високим ризиком її розвитку по можливості слід уникати призначення діуретиків або β-блокаторів. Проведені дослідження показали високу ефективність лозартану не тільки для корекції АТ, але і гіперурикемії, зокрема у пацієнтів із подагрою. З огляду на ефективність, добру переносимість, метаболічну нейтральність, а також безпеку застосування препаратами вибору у пацієнтів з АГ у поєднанні з подагрою є пролонговані антагоністи кальцію (амлодипін, ніфедипін).
Конфлікт інтересів. Автори заявляють про відсутність конфлікту інтересів при підготовці даної статті, при цьому автори не отримували від окремих осіб і організацій фінансової підтримки дослідження, гонорарів та інших форм винагородження.
Інформація про вклад кожного автора: Ханюков О.О. — концепція та дизайн дослідження; Єгудіна Є.Д. — огляд літератури з проблеми, написання тексту; Калашникова О.С. — підготовка ілюстрацій, написання тексту.
Список литературы
1. Maiuolo J. Regulation of uric acid metabolism and excretion / J. Maiuolo, F. Oppedisano, S. Gratteri [et al.] // Int. J. Cardiol. — 2016. — Vol. 213. — P. 8-14.
2. Dalbeth N. Gout / N. Dalbeth, T. Merriman, L. Stamp // Lancet. — 2016. — Vol. 388, № 10055. — P. 2039-2052.
3. Norvik J.V. Overweight modifies the longitudinal association between uric acid and some components of the metabolic syndrome: The Tromsø Study / J.V. Norvik, H.M. Storhaug, K. Ytrehus [et al.] // BMC Cardiovasc. Disord. — 2016. — Vol. 16. — P. 85-98.
4. Lin K.C. The interaction between uric acid level and other risk factors on the development of gout among asymptomatic hyperuricemic men in a prospective study / K.C. Lin, H.Y. Lin, P. Chou // J. Rheumatol. — 2000. — Vol. 27, № 6. — P. 1501-1505.
5. Abeles A.M. Hyperuricemia, gout, and cardiovascular disease: an update / A.M. Abeles // Curr. Rheumatol. Rep. — 2015. — Vol. 17, № 3. — P. 13.
6. Billiet L. Review of Hyperuricemia as New Marker for Metabolic Syndrome / L. Billiet, S. Doaty, J.D. Katz, M.T. Velasquez // ISRN Rheumatol. — 2014. — Р. 1-7.
7. Enomoto A. Roles of organic anion transporters (OATs) and a urate transporter (URAT1) in the pathophysiology of human disease / A. Enomoto, H. Endou // Clin. Exp. Nephrol. — 2005. — Vol. 9(3). — P. 195-205.
8. Feig D.I. Uric acid and cardiovascular risk / D.I. Feig, D.H. Kang, R.J. Johnson // N. Engl. J. Med. — 2008. — Vol. 359, № 17. — P. 1811-1821.
9. PerezRuiz F. Inflammation: a possible mechanism for a causative role of hyperuricemia/gout in cardiovascular disease / F. Perez–Ruiz, M.A. Becker // Curr. Med. Res. Opin. — 2015. — Vol. 31, № 2. — P. 9-14.
10. Glantzounis G.K. Uric acid and oxidative stress / G.K. Glantzounis, E.C. Tsimoyiannis, A.M. Kappas, D.A. Galaris // Curr. Pharm. Des. — 2005. — Vol. 11, № 32. — P. 4145-4151.
11. Kucharz E.J. Chronic inflammationenhanced atherosclerosis: can we consider it as a new clinical syndrome? / E.J. Ku-charz // Med. Hypotheses. — 2012. — Vol. 78, № 3. — P. 396-397.
12. Smith E. The global burden of gout: estimates from the Global Burden of Disease 2010 study / E. Smith, D. Hoy, M. Cross [et al.] // Ann. Rheum. Dis. — 2014. — Vol. 73, № 8. — P 1470-6.
13. Roddy E. Epidemiology of gout / E. Roddy, H.K. Choi // Rheum. Dis. Clin. North Am. — 2014. — Vol. 40, № 2. — P. 155-175.
14. Qiu L. Prevalence of hyperuricemia and its related risk factors in healthy adults from Northern and Northeastern Chinese provinces / L. Qiu, Xq. Cheng, J. Wu [et al.] // BMC Public Health. — 2013. — Vol. 13. — P. 664.
15. Perlstein T.S. Uric acid and the development of hypertension: the normative aging study / T.S. Perlstein, O. Gumieniak, G.H. Williams [et al.] // Hypertension. — 2006. — Vol. 48, № 6. — P. 1031-1036.
16. Syamala S. Association between serum uric acid and prehypertension among US adults / S. Syamala, J. Li, A. Shankar // J. Hypertens. — 2007. — Vol. 25(8). — P. 1583-1589.
17. Kuwabara M. Hyperuricemia, Cardiovascular Disease, and Hypertension / M. Kuwabara // Pulse (Basel). — 2016. — Vol. 3, № 3–4. — P. 242-252.
18. Ruggiero C. Uric acid and dementia in communitydwelling older persons / C. Ruggiero, A. Cherubini, F. Lauretani [et al.] // Dement. Geriatr. Cogn. Disord. — 2009. — Vol. 27, № 4. — P. 382-389.
19. Sautin Y.Y. Uric acid: the oxidant–antioxidant paradox / Y.Y. Sautin, R.J. Johnson // Nucleosides Nucleotides Nucleic Acids. — 2008. — Vol. 27, № 6. — P. 608-619.
20. Ioachimescu A.G. Serum uric acid is an independent predictor of allcause mortality in patients at high risk of cardiovascular disease: a preventive cardiology information system (PreCIS) database cohort study / A.G. Ioachimescu, D.M. Brennan, B.M. Hoar [et al.] // Arthritis Rheum. — 2008. — Vol. 58, № 2. — P. 623-630.
21. Grygiel-Górniak B. Uric acid and obesity-related phenotypes in postmenopausal women / B. Grygiel-Górniak, M. Mosor, J. Marcinkowska [et al.] // Mol. Cell. Biochem. — 2018. — Vol. 443, № 1. — P. 111-119.
22. Puddu P. Relationships among hyperuricemia, endothelial dysfunction and cardiovascular disease: molecular mechanisms and clinical implications / P. Puddu, G.M. Puddu, E. Cravero [et al.] // J. Cardiol. — 2012. — Vol. 59, № 3. — P. 235-242.
23. Mahomed F.A. On chronic Bright’s disease, and its essential symptoms / F.A. Mahomed // Lancet. — 1879. — Vol. 1. — P. 399-401.
24. Haig A. On uric acid and arterial hypertension / A. Haig // BMJ. — 1889. — Vol. 1. — P. 288-91.
25. Huchard H. Allgemeine Betrachtungen über die Arterio-sklerose / H. Huchard // Klin. Med. Berlin. — 1909. — Vol. 5. — P. 1318-21.
26. Mikkelsen W.M. The distribution of serum uric acid values in a population unselected as to gout or hyperuricemia Tecumseh, Michigan 1959–1960 / W.M. Mikkelsen, H.J. Dodge, H. Valkenburg // Am. J. Med. — 1965. — Vol. 39. — P. 242-51.
27. Wallace S.L. Gout and hypertension / S.L. Wallace // Arthr. Rheum. — 1975. — Vol. 18. — P. 721-3.
28. Alper A.B. Jr. Childhood uric acid predicts adult blood pressure: the Bogalusa Heart Study / A.B. Jr Alper, W. Chen, L. Yau [et al.] // Hypertension. — 2005. — Vol. 45. — P. 34-8.
29. Lee J.E. Serum uric acid is associated with microalbuminuria in prehypertension / J.E. Lee, Y.G. Kim, Y.H. Choi [et al.] // Hypertension. — 2006. — Vol. 47, № 5. — P. 962-967.
30. Nagahama K. Hyperuricemia as a predictor of hypertension in a screened cohort in Okinawa, Japan / K. Nagahama, T. Inoue, K. Iseki [et al.] // Hypertens Res. — 2004. — Vol. 27(11). — P. 835-41.
31. Taniguchi Y. Serum uric acid and the risk for hypertension and Type 2 diabetes in Japanese men: The Osaka Health Survey / Y. Taniguchi, T. Hayashi, K. Tsumura [et al.] // J. Hypertens. — 2001. — Vol. 19, № 7. — P. 1209-15.
32. Grayson P. Hyperuricemia and Incident Hypertension: A Systematic Review and Meta-Analysis / P. Grayson, S.Y. Kim, M. LaValley, K.I. Cho Hyon // Arthritis Care Res (Hoboken). — 2011. — Vol. 63, № 1. — P. 102-110.
33. Sundström J. Relations of serum uric acid to longitudinal blood pressure tracking and hypertension incidence / J. Sundström, L. Sullivan, R.B. D’Agostino [et al.] // Hypertension. — 2005. — Vol. 45, № 1. — P. 28-33.
34. Ndrepepa G. Prognostic value of uric acid in patients with acute coronary syndromes / G. Ndrepepa, S. Braun, H.U. Haase [et al.] // Am. J. Cardiol. — 2012. — Vol. 109, № 9. — P. 1260-1265.
35. Fang J. Serum uric acid and cardiovascular mortality: The NHANES I epidemiologic follow-up study, 1971–1992. National Health and Nutrition Examination Survey / J. Fang, M. Alderman // JAMA. — 2000. — Vol. 238. — P. 2404-10.
36. Verdecchia P. Relation between serum uric acid and risk of cardiovascular disease in essential hypertension. The PIUMA study / P. Verdecchia, G. Schillaci, G. Reboldi [et al.] // Hypertension. — 2000. — Vol. 36. — P. 1072-8.
37. Bhole V. Epidemiology of gout in women: fifty-two-year followup of a prospective cohort / V. Bhole, M. de Vera, M.M. Rahman, E. Krishnan, H. Choi // Arthritis Rheum. — 2010. — Vol. 62. — P. 1069-76.
38. Pillinger M.H. Gout and its comorbidities / M.H. Pillinger, D.S. Goldfarb, R.T. Keenan // Bull. NYU Hosp. Jt. Dis. — 2010. — Vol. 68. — P. 199-203.
39. Li Q. The Association between Serum Uric Acid Levels and the Prevalence of Vulnerable Atherosclerotic Carotid Plaque: A Cross-sectional Study / Li Q., Zhou Y., Dong K. [et al.] // Sci Rep. — 2015. — Vol. 5. — P. 1003.
40. Gibson T.J. Hypertension, its treatment, hyperuricemia and gout / T.J. Gibson // Curr. Opin. Rheumatol. — 2013. — Vol. 25, № 2. — P. 217-22.
41. Krishnan E. Gout and risk of acute myocardial infarction / E. Krishnan, J.F. Baker, D.E. Furst et al. // Arthr. Rheum. — 2006. — Vol. 54(8). — P. 2688-96.
42. Choi H.K. Pathogenesis of gout / H.K. Choi, D.B. Mount, A.M. Reginato // Ann. Intern. Med. — 2005. — Vol. 143, № 7. — P. 499-516.
43. Johnson R.J. Is there a pathogenetic role for uric acid in hypertension and cardiovascular and renal disease? / R.J. Johnson, D.N. Kang, D. Feig [et al.] // Hypertension. — 2003. — Vol. 41. — P. 1183-90.
44. Johnson R.J. A unifying pathway for essential hypertension / R.J. Johnson, B. Rodriguez-Iturbe, D.H. Kang [et al.] // Am. J. Hypertens. — 2005. — Vol. 18. — P. 431-40.
45. Nakagawa Т. Hyperuricemia causes glomerular hypertrophy in the rat / T. Nakagawa, M. Mazzali, D.-H. Kang [et al.] // Am. J. Nephrol. — 2003. — Vol. 23. — P. 2-7.
46. Milionis H. Effects of statin treatment on uric acid homeostasis in patients with primary hyperlipidemia / H. Milionis, A. Kakafika // Am. Heart. J. — 2004. — Vol. 148, № 4. — P. 635-640.
47. Mazzali M. Elevated uric acid increases blood pressure in the rat by a novel crystal-independent mechanism / M. Mazzali, J. Hughes, Y.G. Kim [et al.] // Hypertension. — 2001. — Vol. 38, № 5. — P. 1101-6.
48. Mazzali M. Hyperuricemia induces a primary renal arteriolopathy in rats by a blood pressure-independent mechanism / M. Mazzali, J. Kanellis, L. Han [et al.] // Am. J. Physiol. Renal. Physiol. — 2002. — Vol. 282, № 6. — P. 991-7.
49. Puig J.G. Uric acid as a cardiovascular risk factor in arterial hypertension / Puig J.G., L.M. Ruilope // J. Hypertens. — 1999. — Vol. 17. — P. 869-72.
50. Leyva F. Serum Uric Acid as an index of impaired oxidative metabolism in chronic heart failure / F. Leyva, S. Anker, J.W. Swan [et al.] // Eur. Heart J. — 1997. — Vol. 8. — P. 858-65.
51. Johnson R.J. Subtle acquired renal injury as a mechanism of salt-sensitive hypertension / R.J. Johnson, J. Herrera-Acosta, G.F. Schreiner, B. Rodriguez-Iturbe // N. Engl. J. Med. — 2002. — Vol. 346. — P. 913-23.
52. Berni A. Serum uric acid levels and renal damage in hyperuricemic hypertensive patients treated with renin-angiotensin system blockers / A. Berni, M. Boddi, E.B. Fattori [et al.] // Am. J. Hypertens. — 2010. — Vol. 23, № 6. — P. 675-80.
53. Thottam G.E. Gout and Metabolic Syndrome: a Tangled Web / G.E. Thottam, S. Krasnokutsky, M.H. Phillinger [et al.] // Curr. Rheumatol. Rep. — 2017. — Vol. 19(10). — P. 60.
54. Simonenko V.B. Pathogenetic aspects of arterial hypertension in metabolic syndrome / V.B. Simonenho, I.N. Medvedev // Klin. Med. (Mosk.). — 2011. — Vol. 89(1). — P. 49-51.
55. El-Atat F.A. The Relationship between Hyperinsulinemia, Hypertension and Progressive Renal Disease / A.F. El-Atat, S.N. Sameer, I. Samy., J.R. Sowers // JASN. — 2004. — Vol. 15(11). — P. 2816-2827.
56. Tarray R. Role of insulin resistance in essential hypertension / R. Tarray, S Saleem, D. Afroze [et al.] // Cardiovascular Endocrinology & Metabolism. — 2014. — Vol. 3(4). — P. 129-133.
57. Duvnjak L. Hypertension and the metabolic syndrome / L. Duvnjak., B. Tomislav, M. Željko // Diabetologia Croatica. — 2008. — Vol. 37. — P. 4.
58. Babu Giridhara R. Association of obesity with hypertension and type 2 diabetes mellitus in India: A meta-analysis of observational studies / R. Babu Giridhara, G.V.S. Murthy, A. Yamuna [et al.] // World J. Diabetes. — 2018. — Vol. 9(1). — P. 40-52.
59. Thorp A.A., Schlaich M.P. Relevance of Sympathetic Nervous System Activation in Obesity and Metabolic Syndrome / A.A. Thorp, M.P. Schlaich // Journal of Diabetes Research. — 2015. — Vol. 2015. — P. 11.
60. Brent M.E. Insulin resistance and the sympathetic nervous system / M.E. Brent // Current Hypertension Reports. — 2003. — Vol 5(3). — P. 247-254.
61. Wang Q. Inhibiting Insulin-Mediated β2-Adrenergic Receptor Activation Prevents Diabetes-Associated Dysfunction / Q. Wang, Y. Liu, Q. Fu [et al.] // Cardiac. Circulation. — 2017. — Vol. 135. — P. 73-88.
62. Rabmouni K. Obesety-associated hypertension. New insights into mechanisms / K. Rabmouni, M.L.G. Correia, W.G. Hayens [el al.] // Hypertension. — 2005. — Vol. 45. — P. 9-14.
63. Tang L. Hyperuricemia in obese children and adolescents: the relationship with metabolic syndrome / L. Tang, M. Kubota, A. Nagai [et al.] // Pediatr. Rep. — 2010. — Vol. 2(1). — P.e12.
64. Mancia G. Guidelines for the Management of Arterial Hypertension: The Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESC) / G. Mancia, G. de Backer, A. Dominiczak [et al.] // J. Hypertens. — 2007. — Vol. 25(6). — P. 1105-87.
65. Jiang Y. Independent stem cell lineages regulate adipose organogenesis and adipose homeostasis / Y. Jiang, D.C. Berry, W. Tang, J.M. Graff // Cell. Rep. — 2014. — Vol. 9(3). — P. 1007-22.
66. Stern J.H. Adiponectin, Leptin, and Fatty Acids in the Maintenance of Metabolic Homeostasis Through Adipose Tissue Crosstalk Cell / J.H. Stern, J.M. Rutkowski, P.E. Scherer // Metab. — 2016. — Vol. 23(5). — P. 770-784.
67. Belfort R. Dose-Response Effect of Elevated Plasma Free Fatty Acid on Insulin Signaling / R. Belfort, L. Mandarino, S. Kashyap [et al.] // Diabetes. — 2005. — Vol. 54(6). — P. 1640-1648.
68. Chu N.F. Plasma leptin concentrations and obesity in relation to insulin resistance syndrome components among school children in Taiwan-The Taipei Children Heart Study / N.F. Chu, D.J. Wang, S.M. Shieh, E.B. Rimm // Int. J. Obes. Relat. Metab. Disord. — 2000. — Vol. 24(10). — P. 1265-71.
69. Filer J.S. Leptin resistance and obesity / Presented at the 60th Scientific Sessions of the American diabetes association. — Texas: San-Antonio, 2000.
70. Yamagishi S.I. Leptin induces mitochondrial superoxide production and monocyte chemoattractant protein-1 expression in aortic endothelial cells by increasing fatty acid oxidation via protein kinase / S.I. Yamagishi, D. Edelstein, X.L. Du [et al.] // A. J. Biol. Chem. — 2001. — Vol. 276(27). — P. 25096-100.
71. Quehenberger P. Leptin induces endothelin-1 in endothelial cells in vitro / P. Quehenberger, M. Exner, R. Sunder [et al.] // Circ. Res. — 2002. — Vol. 90. — P. 711-8.
72. Shin H.J. Leptin induces hypertrophy via p38 mitogen-activated protein kinase in rat vascular smooth muscle cells / H.J. Shin, J. Oh, S.M. Kang [et al.] // Biochem. Biophys. Res. Commun. — 2005. — Vol. 329. — P. 18-24.
73. Parhami F. Leptin enhances the calcification of vascular cells: artery wall as a target of leptin / F. Parhami, Y. Tintut, A. Ballard [et al.] // Circ. Res. — 2001. — Vol. 88. — P. 954-60.
74. Efstratiadis G. Leptin as a cardiovascular risk factor / G. Efstratiadis, C. Nikolaidou // Hippokratia. — 2007. — Vol. 11(4). — P. 163-170.
75. Söderberg S. High leptin levels are associated with stroke / S. Söderberg, B. Stegmayr, C. Ahlbeck-Glader [et al.] // Cerebrovasc. Dis. — 2003. — Vol. 15(1–2). — P. 63-9.
76. Mathew J. Reduction of cardiovascular risk by regression of elecrocardiographic markers of left ventricular hypertrophy by the angiotensin enzyme inhibitor, ramipril / J. Mathew, P. Sleight, E. Lonn [et al.] // Circulation. — 2001. — Vol. 104. — P. 1615-21.
77. Cuspidi C. Left-ventricular hypertrophy and obesity: a systematic review and meta-analysis of echocardiographic studies / C. Cuspidi, M. Rescaldani, C. Sala, G. Grassi // J. Hypertens. — 2014. — Vol. 32(10. — P. 16-25. doi: 10.1097/HJH.0b013e328364fb58.
78. Kuo C.F., Yu K.H., Luo S.F. et al. Role of uric acid in the link between arterial stiffness and cardiac hypertrophy: a cross-sectional study / C.F. Kuo, K.H. Yu, S.F. Luo // Rheumatology. — Vol. 49(6). — P. 1189-96.
79. Struijker Boudier H.A.J. Hypertension and the microcirculation / In: Kaplan N., ed. Hypertension, microcirculation and end organ damage. — London: Lippincott Williams & Wilkins, 2002. — P. 49-55.
80. Chamorro A. Prognostic Significance of Uric Acid Serum Concentration in Patients with Acute Ischemic Stroke / A. Chamorro, V. Obach, A. Cervera [et al.] // Stroke. — 2002. — Vol. 33(4). — P. 1048-1052.
81. Amaro S. A Pilot Study of Dual Treatment with Recombinant Tissue Plasminogen Activator and Uric Acid in Acute Ischemic Stroke / S. Amaro, D. Soy, V. Obach [et al.] // Stroke. — 2007. — Vol. 38(7). — P. 2173-2175.
82. Duan W. Dietary folate deficiency and elevated homocysteine levels endanger dopaminergic neurons in models of Parkinson’s disease / W. Duan, B. Ladenheim, R.G. Cutler [et al.] // J. Neurochem. — 2002. — Vol. 80(1). — P. 101-110.
83. Imaram W. Radicals in the reaction between peroxynitrite and uric acid identified by electron spin resonance spectroscopy and liquid chromatography mass spectrometry / W. Imaram, C. Gersch, K.M. Kim [et al.] // Free Radic. Biol. Med. — 2010. — Vol. 49(2). — P. 275-281.
84. Ríos María T., Domínguez-Sardiña M., Ayala Diana E. et al. Prevalence and Clinical Characteristics of Isolated-Office and True Resistant Hypertension Determined by Ambulatory Blood Pressure Monitoring / T. Ríos María, M. Domínguez-Sardiña, E. Ayala Diana [et al.] // Journal of Biological and Medical Rhythm Research. — 2013. — Vol. 30(1–2). — P. 207-220.
85. Höcht C. Blood Pressure Variability: Prognostic Value and Therapeutic Implications / C. Höcht // ISRN Hypertension. — 2013. — Vol. 2013. — P. 16.
86. Nagano M. Association of serum uric acid with subsequent arterial stiffness and renal function in normotensive subjects / M. Nagano, M. Takahashi, N. Miyai [et al.] // Hypertens Res. — 2017. — Vol. 40(6). — P. 620-624. doi: 10.1038/hr.2017.10.
87. Торбас О.О. Вплив гіперурикемії на пружно-еластичні властивості артерій у пацієнтів з артеріальною гіпертензією / О.О. Торбас, О.Л. Рековець, Ю.М. Сіренко // Артеріальна гіпертензія. — 2017. — № 4(54). — С. 58-65.
88. Armstrong A.C. LV mass assessed by echocardiography and CMR, cardiovascular outcomes, and medical practice / A.C. Armstrong, S. Gidding, O. Gjesdal [et al.] // J. Am. Coll. Cardiol. Img. — 2012. — Vol. 5. — P. 837-438.
89. Greenland P. 2010 ACCF/AHA guideline for assessment of cardiovascular risk in asymptomatic adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines / P. Greenland, J.S. Alpert, G.A. Beller [et al.] // J. Am. Coll. Cardiol. — 2010. — Vol. 56. — P. e50-103.
90. Okwuosa T.M. Left ventricular hypertrophy and cardiovascular disease risk prediction and reclassification in blacks and whites: the Atherosclerosis Risk in Communities Study / T.M. Okwuosa, E.Z. Soliman, F. Lopez [et al.] // Am. Heart J. — 2015. — Vol. 169. — P. 155-161.
91. Mara A. Diuretic Use, Increased Serum Urate and the Risk of Incident Gout in a Population-based Study of Hypertensive Adults: the Atherosclerosis Risk in the Communities Cohort / A. Mara, W. Janet, N. Alan [et al.] // Arthritis Rheum. — 2012. — Vol. 64(1). — P. 121-129.
92. Reyes A.J. Cardiovascular drugs and serum uric acid / A.J. Reyes // Cardiovasc. Drugs. Ther. — 2003. — Vol. 17(5–6). — P. 397-414.
93. Hunter D.J. Recent diuretic use and the risk of recurrent gout attacks: the online case-crossover gout study / D.J. Hunter, M. York, C.E. Chaisson [et al.] // J. Rheumatol. — 2006. — Vol. 33(7). — P. 1341-5.
94. Taniguchi Y. Serum uric acid and the risk for hypertension and Type 2 diabetes in Japanese men: The Osaka Health Survey / Y. Taniguchi, T. Hayashi, K. Tsumura [et al.] // J. Hypertens. — 2001. — Vol. 19(7). — P. 1209-15.
95. El-Sheikh A.A.K. Effect of hypouricaemic and hyperuricaemic drugs on the renal urate efflux transporter, multidrug resistance protein 4 / A.A.K. El-Sheikh, J.J.M.W. van den Heuvel, J.B. Koenderink, F.G.M. Russel // Br. J. Pharmacol. — 2008. — Vol. 155(7). — P. 1066-1075.
96. Ruilope L.M. Uric acid and other renal function parameters in patients with stable angina pectoris participating in the ACTION trial: impact of nifedipine GITS (gastro-intestinal therapeutic system) and relation to outcome / L.M. Ruilope, B.A. Kirwan, S. de Brouwer [et al.] // J. Hypertens. — 2007. — Vol. 25(8). — P. 1711-8.
97. Hoieggen A. The impact of serum uric acid on cardiovascular outcomes in the LIFE study / A. Hoieggen, M.H. Alderman, S.E. Kjeldsen [et al.] // Kidney Int. — 2004. — Vol. 65. — P. 1041-9.
98. Nishida Y. Comparative Effect of Calcium Channel Blockers on Glomerular Function in Hypertensive Patients with Diabetes Mellitus / Y. Nishida, Y. Takahashi, K. Tezuka [et al.] // Drugs. RD. — 2017. — Vol. 17(3). — P. 403-412.
99. Chanard J. Amlodipine reduces cyclosporin-induced hyperuricaemia in hypertensive renal transplant recipients / J. Chanard, O. Toupance, S. Lavaud [et al.] // Nephrol. Dial. Transplant. — 2003. — Vol. 18(10). — P. 2147-53.
100. Wahda B.A.Y., Yasir Y.T.A. Serum uric acid level and renal function tests in hypertensive patients treated by captopril / B.A.Y. Wahda, Y.T.A. Yasir // Iraqi Journal of Pharmacy. — 2013. — Vol. 13(2). — P. 1-10.
101. Li H. Effects of combined enalapril and folic acid therapy on the serum uric acid levels in hypertensive patients: a multicenter, randomized, double-blind, parallel-controlled clinical trial / H.Li, X. Qin, D. Xie [et al.] // Intern. Med. — 2015. — Vol. 54(1). — P. 17-24.
102. Myers M.G. Fixed low-dose combination therapy in hypertension — a dose response study of perindopril and indapamide / M.G. Myers, R. Asmar, F.H. Leenen, M. Safar // J. Hypertens. — 2000. — Vol. 18(3). — P. 317-25.
103. Dahlof B. Cardiovascular morbidity and mortality in the losartan intervention for endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol / B. Dahlof, R.B. Devereux, S.E. Kjeldsen [et al.] // Lancet. — 2002. — Vol. 359. — P. 995-1003.
104. Shahinfar S. Safety of losartan in hypertensive patients with thiazide-induced hyperuricemia / S. Shahinfar, R.L. Simpson, A.D. Carides [et al.] // Kidney Int. — 1999. — Vol. 56. — P. 1879-85.
105. González-Ortiz M. Effect of valsartan on renal handling of uric acid in healthy subjects/M. González-Ortiz, J.M. Mora-Martínez, E. Martínez-Abundis, B.R. Balcázar-Muñoz // J. Nephrol. — 2000. — Vol. 13(2). — P. 126-8.
106. Manolis A.J. Effects of losartan and candesartan monotherapy and losartan/hydrochlorothiazide combination therapy in patients with mild to moderate hypertension. Losartan Trial Investigators / A.J. Manolis, E. Grossman, B. Jelakovic [et al.] // Clin. Ther. — 2000. — Vol. 22(10). — P. 1186-203.
107. Burgess E.D., Buckley S. Acute natriuretic effect of telmisartan in hypertensive patients / E.D. Burgess, S. Buckley // Am. J. Hypertens. — 2000. — Vol. 13. — P. 183A.
108. Würzner G. Comparative effects of losartan and irbesartan on serum uric acid in hypertensive patients with hyperuricaemia and gout / G. Würzner, J.C. Gerster, A. Chiolero [et al.] // J. Hypertens. — 2001. — Vol. 19(10). — P. 1855-60.
109. Rubio-Guerra A.F. Effect of losartan combined with amlodipine or with a thiazide on uric acid levels in hypertensive patients / A.F. Rubio-Guerra, K.G. Ana, I.E. Cesar, A.S. Juan, B.D. Montserrat // Ther. Adv. Cardiovasc. Dis. — 2017. — Vol. 11(2). — P. 57-62.
110. Dincer E. Asymptomatic hyperuricemia: To treat or not to treat / E. Dincer, A. Dincer, D. Levinson // Cleveland Clinic. Journal of Medicine. — 2002. — Vol. 69(8). — P. 594-597.
111. Kelkar A. Allopurinol as a cardiovascular drug / A. Kelkar, A. Kuo, W.H. Frishman // Cardiol. Rev. — 2011. — Vol. 19(6). — P. 265-271.
112. Stamp L.K. Starting dose is a risk factor for allopurinol hypersensitivity syndrome: a proposed safe starting dose of allopurinol / L.K. Stamp, W.J. Taylor, P.B. Jones [et al.] // Arthritis Rheum. — 2012. — Vol. 64(8). — P. 2529-2536.
113. Seth R. Allopurinol for chronic gout / R. Seth, A. Kydd, R. Buchbinder [et al.] // Cochrane Database of Systematic Reviews. — 2014.
114. Luk A.J. Allopurinol and mortality in hyperuricaemic patients / A.J. Luk, G.P. Levin, E.E. Moore [et al.] // Rheumatology (Oxford). — 2009. — Vol. 48(7). — P. 804-806.
115. Dubreuil M. Allopurinol initiation and allcause mortality in the general population / M. Dubreuil, Y. Zhu, Y. Zhang [et al.] // Ann. Rheum. Dis. — 2015. — Vol. 74(7). — P. 1368-1372.
116. Deng G. Effects of Allopurinol on Arterial Stiffness: A MetaAnalysis of Randomized Controlled Trials / G. Deng, Z Qiu, D. Li [et al.] // Med. Sci Monit. — 2016. — Vol. 22. — P. 1389-1397.
117. Bruce S.P. Febuxostat: a selective xanthine oxidase inhibitor for the treatment of hyperuricemia and gout / S.P. Bruce // Ann. Pharmacother. — 2006. — Vol. 40(12). — P. 2187-2194.

/38.jpg)