
Журнал «» №3-4 (65-66), 2019
Вернуться к номеру
Зв’язок синдрому обструктивного апное сну та легеневої гіпертензії (огляд літератури)
Авторы: Крушинська Н.А.(1), Сіренко Ю.М.(2)
(1) — Національний медичний університет імені О.О. Богомольця, м. Київ, Україна
(2) — ДУ «ННЦ «Інститут кардіології імені акад. М.Д. Стражеска» НАМН України, м. Київ, Україна
Рубрики: Кардиология
Разделы: Справочник специалиста
Версия для печати
У реальній клінічній практиці найчастіше зустрічається легенева гіпертензія (ЛГ) внаслідок хронічних захворювань дихальної системи та/або гіпоксії, яку визначають як ЛГ ІІІ групи за класифікацією Всесвітньої організації охорони здоров’я. Однією з найчастіших причин такої ЛГ є синдром обструктивного апное сну. Метою даного огляду є аналіз літературних даних щодо зв’язку синдрому обструктивного апное сну та ЛГ: клінічна значущість, патогенетичні механізми, обґрунтування необхідності ранньої діагностики та підходи до ведення таких пацієнтів.
В реальной клинической практике наиболее часто встречается легочная гипертензия (ЛГ) вследствие хронических заболеваний дыхательной системы и/или гипоксии, которую обозначают как ЛГ ІІІ группы по классификации Всемирной организации здравоохранения. Одной из самых частых причин такой ЛГ является синдром обструктивного апноэ сна. Целью данного обзора является анализ литературных данных о связи синдрома обструктивного апноэ сна и ЛГ: клиническая значимость, патогенетические механизмы, обоснование необходимости ранней диагностики и подходы к ведению таких пациентов.
Pulmonary hypertension secondary to chronic respiratory diseases and/or hypoxia is the most frequent in real clinical practice. It is classified as World Health Organization group III pulmonary hypertension. One of the most frequent causes of pulmonary hypertension is obstructive sleep apnea. The purpose of this review is to analyze the literature on the correlation between obstructive sleep apnea syndrome and pulmonary hypertension: clinical significance, pathogenesis, proving the necessity of early diagnosis and summarizing approaches to the management of these patients.
легенева гіпертензія; синдром обструктивного апное сну; СРАР-терапія; огляд
легочная гипертензия; синдром обструктивного апноэ сна; СРАР-терапия; обзор
pulmonary hypertension; obstructive sleep apnea; continuous positive airway pressure therapy; review
Вступ
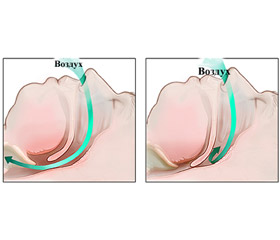
Синдром обструктивного апное сну (СОАС) — стан, при якому в пацієнта виникають численні зупинки дихання, що повторюються, внаслідок повного перекриття (апное) або часткового звуження (гіпопное) дихальних шляхів під час сну на рівні глотки і припинення легеневої вентиляції при збережених дихальних зусиллях [1].
Легенева гіпертензія (ЛГ) — це гемодинамічний та патофізіологічний стан, що характеризується підвищенням середнього тиску в легеневій артерії (ЛА) ≥ 25 мм рт.ст. у спокої й оцінюється за даними катетеризації правих відділів серця (КПС) [25].
Згідно з класифікацією Всесвітньої організації охорони здоров’я (ВООЗ), виділяють 5 груп ЛГ. ЛГ, що розвивається внаслідок СОАС, належить до ІІІ групи — ЛГ, яка виникла у зв’язку із захворюваннями легень та/або гіпоксією (так звана гіпоксична легенева гіпертензія) [19]. На відміну від легеневої артеріальної гіпертензії (ЛАГ, група І ВООЗ), яка є рідкісним захворюванням, ЛГ ІІІ групи у клінічній практиці зустрічається набагато частіше, вивчена набагато менше і відзначається браком ефективного лікування.
ЛГ ІІІ групи при катетеризації правих відділів серця характеризується рисами прекапілярної ЛГ (середній тиск у ЛА > 25 мм рт.ст., тиск заклинювання ЛА (ТЗЛА) < 15 мм рт.ст. із нормальним або зниженим серцевим викидом). Виникає така ЛГ вторинно до хронічних обструктивних захворювань легень (ХОЗЛ), інтерстиційних хвороб легень, захворювань легень із поєднаним обструктивним та рестриктивним типом, порушень дихання під час сну, гіповентиляційних розладів, хронічної гіпоксії або порушень розвитку легень [18].
Актуальність
Актуальність проблеми зумовлена значною частотою СОАС середнього та тяжкого ступенів, що, за останніми дослідженнями, в загальній популяції досягає 50 % у чоловіків та 23 % у жінок [23], при цьому він не діагностується у 60–92 % хворих [17, 48]. СОАС, особливо тяжкий, супроводжується численними серцево–судинними та метаболічними ускладненнями, одним з яких є розвиток ЛГ.
Частота ЛГ при СОАС становить 27–30 % за відсутності інших відомих серцево–легеневих захворювань [6, 46]. З іншого боку, хворі з ЛГ різної етіології мають високу частоту СОАС, що погіршує якість життя таких пацієнтів. Так, у пацієнтів із ЛАГ та хронічною тромбоемболічною ЛГ (ХТЕЛГ) з ІІ–ІІІ функціональним класом за ВООЗ без ХОЗЛ та дисфункції лівого шлуночка (ЛШ) частота СОАС досягає 89 % [28].
Зазвичай СОАС призводить до незначного/помірного підвищення тиску в ЛА, особливо в осіб без захворювань легень. Ці зміни більш значущі в пацієнтів із денною гіпоксемією, яка відмічається при синдромі ожиріння–гіповентиляції, ХОЗЛ або тяжких деформаціях грудної клітки. Загалом хворі із СОАС та ЛГ частіше мають ожиріння, менші об’єм форсованого видиху за 1 с (ОФВ1), життєву ємність легень (ЖЄЛ), нижчий парціальний тиск кисню (PaO2), вищий парціальний тиск вуглекислого газу (PaСO2) та тяжчі нічні гіпоксемії [7].
Загалом ризик ЛГ вищий у пацієнтів із СОАС у поєднанні з ХОЗЛ, або тяжким ожирінням, або з комбінацією обох з них. При цьому немає кореляції між тяжкістю СОАС, вираженою індексом апное–гіпопное (ІАГ), і наявністю та ступенем ЛГ.
Клінічно ЛГ проявляється задишкою при фізичних навантаженнях, слабкістю, болем у грудях, синкопе, серцебиттям та/або набряками нижніх кінцівок. Однак, якщо ЛГ розвивається як ускладнення СОАС, симптоми можуть бути не виражені через ожиріння, низький рівень фізичної активності або можуть маскуватись іншими порушеннями з боку серцево–судинної системи, які часто наявні у хворих із СОАС.
Підтвердження ЛГ у хворих із СОАС важливе з кількох причин:
1. СОАС асоціюється з вищою смертністю у пацієнтів із ЛГ порівняно з пацієнтами без ЛГ [40] та нижчою якістю життя хворих [29]. Загалом розвиток ЛГ у пацієнтів із СОАС свідчить про несприятливий прогноз.
2. Такі хворі потребують спеціального лікування СОАС.
3. При веденні пацієнтів із ЛГ та СОАС слід враховувати можливість інших механізмів розвитку ЛГ, які потребують іншої тактики лікування.
Крім того, наявність СОАС є одним із факторів, які спричиняють затримку діагностики ЛАГ [13].
Саме тому пацієнтів із СОАС слід обстежувати на предмет ЛГ і, навпаки, хворих із діагностованою ЛГ необхідно обстежувати на наявність СОАС.
Патофізіологічні аспекти
Нормальний сон забезпечує сприятливий для серцево–судинної системи період низького фізіологічного навантаження. Так, упродовж фази сну, нешвидкого руху очей (NREM), зменшується симпатична та підвищується парасимпатична активність зі зниженням артеріального тиску (АТ) > 20 %, частоти серцевих скорочень (ЧСС), серцевого викиду (на 10 %) та системного судинного опору, зниженням вентиляції та РаO2 і одночасним підвищенням парасимпатичного тонусу та РаСO2.
Фаза сну швидкого руху очей (REM) характеризується підвищенням симпатичної активності з лабільними ЧСС і АТ, аналогічними періоду неспання, з підвищенням тиску також в ЛА порівняно з NREM–фазою, але ці зміни в здорових осіб не є значущими. Під час REM–фази сну на респіраторну стимуляцію впливають також супутні стани, і пригнічення тонусу м’язів верхніх дихальних шляхів та допоміжних м’язів, зумовлюючи нерегулярне дихання, може погіршувати гіпоксемію та гіперкапнію.
Приблизно 80 % часу сну проводиться в NREM–фазі та 20 % — в REM–фазі.
СОАС характеризується повторними епізодами апное та гіпопное з інтермітуючими гіперкапнією та гіпоксією, підвищенням симпатичної активності та змінами барорецепторної рефлекторної відповіді.
Гострі ефекти альвеолярної гіпоксії з гіпоксичною вазоконстрикцією визначають тривалі впливи (легеневе судинне ремоделювання), що призводить до підвищення резистентності легеневих судин та легеневого тиску. Пороговий рівень гіпоксемії, нижче якого починає підвищуватись тиск в легеневій артерії (ТЛА), становить 55–60 мм рт.ст., що еквівалентне сатурації (SpO2) 88–90 %.
Епізоди апное/гіпопное при СОАС із компенсаторними гіперпное асоціюються з такими серцево–судинними ускладненнями:
1. Змінами газового складу крові з інтермітуючими гіпоксіями/реоксигенаціями та коливаннями РаСО2. Легенева вазоконстрикція є прямою відповіддю на альвеолярну гіпоксію як фізіологічна спроба мінімізувати вентиляційно–перфузійну невідповідність. Залежно від ступеня альвеолярної гіпоксії легеневий судинний опір може зростати більше ніж на 300 % [9]. Епізоди гіпоксії та швидкі реоксигенації в кінці епізоду апное/гіпопное призводять до продукції вільних радикалів, наслідком чого є активація генів окислювально–відновлювальних процесів із оксидативним стресом та активація запального каскаду вазоактивних факторів, таких як серотонін [33], ангіопоетин–1, ендотелін–1 [57], НАДФ–оксидаза [34], що є стимулами вазоконстрикції та клітинної проліферації, і зменшення продукції вазодилататора оксид азоту [56]. Повторні гіпоксемії призводять до повторних підвищень тиску в ЛА, але ЛГ на сьогодні розвивається приблизно в 1 з 5 пацієнтів.
Гіперкапнія та ацидоз, які незалежно спричинюють легеневу вазоконстрикцію, також посилюють ефект гіпоксичної легеневої вазоконстрикції.
2. Зниженням парасимпатичної та підвищенням симпатичної активності. Періодичні підвищення симпатичної активності з вазоконстрикцією та гіпертензією супроводжують епізоди апное з найнижчим АТ посередині цих епізодів. АТ після цього поступово зростає з раптовим підвищенням під час відновлення дихання. Періоди апное тривалістю більше 35 секунд характеризуються зменшенням серцевого викиду більше 33 %, але він зростає до 15 % більше вихідного при відновленні дихання [21, 38, 53]. Підвищений системний АТ та знижений серцевий викид зумовлюють збільшення судинного опору з вазоконстрикцією. У відповідь на гіпоксію та гіперкапнію на початку дихання також різко підвищується тиск у ЛА [49, 51]. Значні коливання системного АТ при СОАС зумовлюють виражені коливання діаметра судин та ендотеліальну дисфункцію [31, 52] із надпродукцією індукованих гіпоксією факторів, таких як прозапальний транскрипційний фактор NF–κB та тумор–некротизуючий фактор α, що спричиняють запальний процес [45]. Це призводить до підвищення ТЛА навіть у денний час за патофізіологічними механізмами, подібними до системної артеріальної гіпертензії (АГ) при СОАС.
3. Значними коливаннями негативного внутрішньогрудного тиску. Визначною рисою СОАС є маневр Mueller — вдих при закритих дихальних шляхах, який може створювати негативний внутрішньогрудний тиск до –80 см вод.ст. Змінені геометрія серця та тиск наповнення камер можуть підвищувати трансмуральний тиск ЛШ та післянавантаження, тоді як релаксація ЛШ порушується внаслідок підвищеного негативного внутрішньогрудного тиску, що погіршує наповнення ЛШ. Це зменшує ударний об’єм та серцевий викид, а негативний внутрішньогрудний тиск розтягує стінку аорти й активує інтрамуральні барорецептори з періодичним пригніченням симпатичного викиду з кожним маневром Mueller. Підвищене венозне повернення при відновленні дихання зумовлює розтягнення правого шлуночка (ПШ) та зміщення міжшлуночкової перегородки ліворуч, порушуючи діастолічне наповнення ЛШ та його розтяжність із підвищенням легеневого венозного тиску з посткапілярною ЛГ [50, 55].
Патологічні зміни при ЛГ внаслідок гіпоксії при СОАС включають гіпертрофію медії та обструктивну проліферацію інтими дистальних легеневих артерій різної тяжкості аж до повної оклюзії цих судин, що призводить до значного підвищення легеневого судинного опору та погіршує проходження крові крізь легені. Головними факторами в цьому процесі є гіпоксична вазоконстрикція, механічні зміни внаслідок перерозтягнення легень, втрата капілярів та запалення з дисбалансом між ендотелійактивними факторами, відповідальними за вазоконстрикцію та вазодилатацію.
З часом тяжкий СОАС може призвести до гіпертрофії ПШ з денною ЛГ та правошлуночковою недостатністю [2, 10], причому при тяжкому СОАС гіпертрофія ПШ більша у порівнянні із легким СОАС.
З іншого боку, СОАС є незалежним фактором ризику венозних тромбоемболічних подій через підвищену активацію тромбоцитів та гіперкоагуляцію [15, 35]. У дослідженні M. Arzt та ін. СОАС частіше реєструвався в пацієнтів із тромбозом глибоких вен та/або гострим легеневим емболізмом, ніж у контрольній групі [5]. Таким чином, фактори ризику венозного тромбоемболізму є факторами ризику хронічної тромбоемболічної ЛГ (IV група ЛГ за класифікацією ВООЗ) [12], що визначає високу поширеність СОАС у пацієнтів із ХТЕЛГ, яка досягає більше 50 %. У такому разі СОАС може погіршувати стан пацієнтів із ХТЕЛГ, що відображається вищим рівнем гемоглобіну, нижчою SpO2 та гіршим функціональним класом ЛГ за ВООЗ внаслідок несприятливого впливу на функцію обох шлуночків, вазоконстрикції та ремоделювання легеневого судинного русла [59].
З іншого боку, затримка рідини при декоменсованій ХТЕЛГ збільшує перифарингеальний набряк під час сну, сприяючи більш значному звуженню верхніх дихальних шляхів [58] та, відповідно, погіршенню СОАС.
Іншим можливим механізмом розвитку ЛГ при СОАС є ЛГ внаслідок ураження лівих відділів серця (група ІІ ЛГ за класифікацією ВООЗ) зі збереженою або зниженою фракцією викиду (ФВ) лівого шлуночка. Як наслідок частої денної гіпертензії та інтермітуючих коливань АТ відповідно до респіраторних подій упродовж сну пацієнти із СОАС мають високу частоту гіпертрофії [22] та дисфункції ЛШ [11, 14, 20]. Так, поширеність ЛГ (визначена як систолічний ТЛА > 35 мм рт.ст. при допплер–ЕхоКГ) при серцевій недостатності (СН) зі збереженою ФВ ЛШ становила 83 %, і її наявність була незалежним предиктором смертності таких пацієнтів [32]. Початкове пасивне підвищення легеневого об’єму крові вторинно щодо підвищеного гідростатичного тиску внаслідок СН призводить до посткапілярної ЛГ із нормальним транспульмональним градієнтом тиску і, відповідно, відносно нормальним легеневим судинним опором.
Так, M. Hetzel та ін. дослідили 49 пацієнтів із СОАС та нормальними функціональними тестами легень і виявили 6 (12 %) пацієнтів із ЛГ у спокої, і ще 39 пацієнтів мали підвищений ТЛА при фізичних навантаженнях, визначений як середній тиск у ЛА > 20 мм рт.ст. за даними КПС. У жодного пацієнта не було підвищеної резистентності легеневих судин, але більшість хворих (64 %) мали значне підвищення ТЗЛА, що вказує на ураження лівих відділів серця як механізм розвитку ЛГ [24].
Тривале підвищення тиску наповнення ЛШ спричиняє розвиток легеневої венозної гіпертензії, поєднання якої з гіпоксичною вазоконстрикцією, продукцією медіаторів запалення призводить до проліферації судинних клітин та судинного ремоделювання, наслідком чого є ЛГ. Ці зміни тотожні змінам, які спостерігаються при інших формах ЛГ, що свідчить про подібність патофізіологічних механізмів.
З іншого боку, порушення дихання під час сну можуть бути вторинними щодо низького серцевого викиду, характерного для пацієнтів із ЛАГ, що асоціюється з правошлуночковою СН та призводить до нестабільності вентиляційного контролю з розвитком нічного апное за механізмами, як при лівошлуночковій недостатності, з диханням Чейна — Стокса/центральним апное [41] та нічною нестабільністю дихання внаслідок рострального перерозподілу рідини в нічний час, як і у пацієнтів із системною АГ [16].
Так, у дослідженні M. Minic та ін. [41] було показано, що 71 % хворих із ЛАГ І групи за ВООЗ мали порушення дихання уві сні: 56 % пацієнтів мали СОАС та 44 % — центральне апное сну, що значно більше, ніж у загальній популяції. Але ніяких відмінностей у серцево–легеневій гемодинаміці та виживанні пацієнтів з порушеннями дихання уві сні та без них у цих пацієнтів виявлено не було.
Таким чином, СОАС може бути причинним фактором, причому за різними механізмами, або супутнім станом щодо легеневої гіпертензії, що в кожного пацієнта потребує подальшого дослідження.
Ведення пацієнтів із СОАС та ЛГ
Ведення пацієнтів із підозрюваною ЛГ потребує низки досліджень для підтвердження діагнозу, що найповніше можливе в мультидисциплінарних центрах ЛГ. Дослідженням першої лінії в діагностиці ЛГ є допплер–ЕхоКГ з оцінкою гемодинаміки правих відділів серця та визначенням тиску в ЛА, що при підозрі на ЛГ є скринінговою методикою. Але точний діагноз ЛГ вимагає катетеризації правих відділів серця та додаткових обстежень, спрямованих на оцінку функціонального стану пацієнтів та пошук можливої причини підвищення тиску в ЛА.
Чіткі показання для проведення КПС при ЛГ та СОАС у чинних настановах не прописані. Щодо загальних рекомендацій, то при веденні пацієнтів із ЛГ ІІІ групи КПС рутинно не рекомендована, окрім спеціальних терапевтичних показань, таких як: 1) точний діагноз або виключення ЛГ у пацієнтів, що є кандидатами на хірургічне лікування; 2) пі–
дозрювана ЛАГ або ХТЕЛГ; 3) епізоди правошлуночкової СН та 4) непереконливі дані ЕхоКГ при значній підозрі або потенційна терапевтична користь.
Відповідно до Настанови з діагностики та лікування ЛГ [19] при веденні пацієнтів із ЛГ необхідні виявлення та лікування станів, які можуть призвести до її розвитку (рівень доказів ІС).
Зараз немає формальних настанов для рутинного сомнологічного обстеження пацієнтів із ЛГ, однак, зважаючи на значну частоту нічних гіпоксемій та апное при ЛГ [28], при клінічній підозрі на СОАС пацієнтів із ЛГ слід обстежувати за допомогою цілонічної оксиметрії або полісомнографії [19] і лікувати СОАС із метою обмеження його несприятливих ефектів на легеневі судини [37].
Але, оскільки СОАС найчастіше спричиняє ЛГ легкого/помірного ступеня, за наявності в пацієнта із СОАС тяжкої ЛГ необхідне комплексне діагностичне обстеження для виявлення іншої можливої причини ЛГ.
При виявленні ЛГ ІІІ групи перший крок у первинному веденні таких хворих — оптимізація первинного захворювання відповідно до клінічних настанов. В останньому оновленні настанови з лікування легеневої артеріальної гіпертензії в дорослих також наголошується на необхідності агресивного лікування причин ЛГ, таких як апное сну та АГ [30].
Лікування ЛГ, асоційованої із СОАС, спрямоване на запобігання періодичним гіпоксіям методом терапії постійним позитивним тиском у дихальних шляхах (СРАР) (із додатковою оксигенотерапією або без неї), або, рідше, хірургічним методом, або з використанням ротових пристроїв [37].
Дані літератури щодо ефективності СРАР у лікуванні ЛГ при СОАС суперечливі. Але переважна кількість досліджень вказують на зниження ТЛА при лікуванні СОАС методом СРАР.
Так, у проспективному дослідженні 20 пацієнтів із СОАС (середній ІАГ — 48,6 ± 5,2 події/год), 4 місяці лікування методом СРАР знизило середній ТЛА з 16,8 ± 1,2 мм рт.ст. до 13,9 ± 0,6 мм рт.ст. (P < 0,05). Найбільший ефект спостерігався в 5 осіб, які мали ЛГ на початку. Зниження ЛГ пояснювалось зменшенням легеневого судинного опору та судинозвужувальної відповіді на гіпоксичні стимули [47].
У рандомізованому плацебо–контрольованому перехресному дослідженні М. Arias із колегами [4] при дослідженні 10 пацієнтів із СОАС та ЛГ (систолічний ТЛА > 30 мм рт.ст. за даними ЕхоКГ) без діагностованого серцевого або легеневого захворювання (середній ІАГ — 44,1 ± 29,3 події/год) ефективна СРАР упродовж 12 тижнів сприяла достовірному зниженню систолічного ТЛА (з 28,9 ± 8,6 мм рт.ст. до 24,0 ± 5,8 мм рт.ст., Р < 0,0001), і зниження було більшим у пацієнтів із СОАС, або діастолічною дисфункцією (ДД) ЛШ (зміна 7,3 ± 3,3 мм рт.ст. проти 1,6 ± 1,8 мм рт.ст. у пацієнтів без ДД ЛШ, Р < 0,001), або наявністю ЛГ на початку дослідження (зміна 8,5 ± 2,8 мм рт.ст. проти 2,6 ± 2,8 мм рт.ст. без ЛГ початково; P < 0,001).
Недоліком цих досліджень є те, що вони не включали дані катетеризації правих відділів серця, тому неможливо визначити, покращання легеневої гемодинаміки було наслідком покращання прекапілярної ЛГ або функції ЛШ.
У дослідженні M. Alchanatis та ін. вивчали вплив СРАР у 6 пацієнтів із СОАС та ЛГ, діагностованою за ЕхоКГ та підтвердженою при катетеризації правих відділів серця, з нормальним ТЗЛА. Після 6 місяців СРАР відмічалося достовірне зниження середнього ТЛА з 25,6 ± 4,0 мм рт.ст. до 19,5 ± 1,6 мм рт.ст. (P < 0,001) [3].
У дослідженні M. Marvisi та ін. СРАР–терапія у 17 хворих із СОАС та ЛГ (систолічний ТЛА — 39,8 ± ± 4,1 мм рт.ст. за ЕхоКГ) без супутніх серцево–судинних захворювань та ХОЗЛ упродовж 9 місяців спостереження супроводжувалась зниженням ТЛА до 27,1 ± 4,0, 25,2 ± 3,1 та 22,2 ± 3,0 мм рт.ст. (95% ДІ 15,09–20,11; Р < 0,001) через 3, 6 та 9 місяців лікування відповідно [36].
За даними метааналізу T. Imran, що включав 222 пацієнти із СОАС та ЛГ, було виявлено, що СРАР–терапія асоціювалась зі зниженням тиску в ЛА на 13,3 мм рт.ст. (95% ДІ 12,7–14,0) [26].
Щодо механізмів позитивного впливу СРАР–терапії при ЛГ отримані дані щодо покращання фракції викиду ПШ, за даними ядерної вентрикулографії, з 30,0 ± 3,0 % до 39,0 ± 3,0 % (Р = 0,01) при лікуванні упродовж 6–24 місяців [42], зниження активності симпатичної нервової системи та зменшення рівнів прозапальних медіаторів, таких як інтерлейкін–6, туморнекротизуючий фактор α та С–реактивний протеїн [8]. Є дані, що СРАР–терапія здатна достатньо швидко нормалізувати рівні таких вазоактивних медіаторів, як ендотелін–1, що підвищується при СОАС, та нітриту азоту, який при СОАС знижується [27, 43], які впливають на судинний легеневий тонус та проліферацію гладком’язових клітин. Відновлення балансу між цими вазоактивними медіаторами при СРАР–терапії відбувається завдяки усуненню нічної гіпоксемії та коливань симпатичного тонусу, покращанню діастолічної релаксації ЛШ, зменшенню перевантаження ЛШ.
Причому вплив СРАР–терапії на ступінь зниження ТЛА залежить від тривалості терапії, коли змінюється співвідношення між функціональними змінами тонусу легеневих судин та структурним ремоделюванням. Так, при короткочасній терапії можуть проявлятись лише функціональні ефекти на артеріальний тонус.
Але незважаючи на такі дані, клінічне значення СРАР–терапії при ЛГ та СОАС залишається невизначеним.
Також при веденні пацієнтів із СОАС та ЛГ можливе застосування кисневої терапії під час сну, особливо у хворих, які не толерантні до СРАР–терапії, що покращувала переносимість фізичних навантажень у дослідженні S. Ulrich та ін. [54], але довгострокова ефективність цієї стратегії в зменшенні ЛГ невідома.
Щодо медикаментозної терапії, то специфічні лікарські препарати для зниження ТЛА не досліджувались у пацієнтів із ЛГ та СОАС, і їх рутинне використання не показане. Але вони можуть використовуватись при прогресуванні ЛГ, незважаючи на адекватну терапію СОАС [37].
Діуретики часто використовуються для нормалізації внутрішньосудинного об’єму в пацієнтів із ЛГ ІІІ групи із затримкою рідини, вторинною щодо правошлуночкової недостатності [19]. Хоча немає рандомізованих клінічних досліджень, що підтримують цю практику, емпіричний клінічний досвід свідчить, що пацієнти отримують симптоматичне покращання при декомпенсованій правошлуночковій недостатності.
Щодо антикоагулянтної терапії її рутинне застосування не рекомендоване в пацієнтів із ЛГ ІІІ групи, окрім специфічних показань в окремих хворих.
Оскільки пневмонія спричиняє більше ніж 7 % смертей хворих із ЛАГ [18], вакцинація від грипу та пневмокока є загальною рекомендацією для усіх груп хворих із ЛГ, включаючи хворих із ЛГ гіпо–ксичного походження.
Також пацієнти із СОАС та ЛГ, ймовірно, матимуть користь від легеневої реабілітації, враховуючи, що фізичні вправи при ЛГ покращують функцію скелетних м’язів, якість життя та толерантність до фізичних навантажень [39, 44]. Ці дані можуть бути використані в пацієнтів із ЛГ внаслідок захворювань легень або гіпоксемії, у яких за можливості необхідно проводити легеневу реабілітацію.
Висновки
1. ЛГ при СОАС найчастіше асоціюється з незначним або помірним підвищенням середнього тиску в ЛА.
2. Розвиток ЛГ при СОАС асоціюється з гіршим функціональним станом та несприятливим прогнозом.
3. Лікування ЛГ при СОАС має бути спрямоване на нормалізацію основного стану в поєднанні з кисневою терапією (за показаннями), діуретиками, легеневою реабілітацією та вакцинацією.
4. Дані щодо застосування судинорозширювальних препаратів при ЛГ ІІІ групи зараз обмежені, і рутинно вони не показані.
5. Лікування СОАС повністю не усуває ЛГ, що потребує подальшого дообстеження для виявлення інших можливих причин й оцінки потреби в фармакологічній терапії.
6. Необхідні подальші дослідження для визначення ефективності медикаментозної терапії в лікуванні ЛГ при СОАС.
Конфлікт інтересів. Автори заявляють про відсутність конфлікту інтересів при підготовці даної статті.
1. Наказ МОЗ України № 384 від 24.05.2012 «Про затвердження та впровадження медико–технологічних документів зі стандартизації медичної допомоги при артеріальній гіпертензії» [Електронний ресурс]. 2012. Режим доступу до ресурсу: https://www.moz.gov.ua/portal/dn_20120524_384.html.
2. Ahmed Q., Chung–Park M., Tomashefski J.F. Cardiopulmonary pathology in patients with sleep apnea/obesity hypoventilation syndrome. Human Pathology. 1997. 28(3). P. 264–269.
3. Alchanatis M., Tourkohoriti G., Kakouros S. et al. Daytime pulmonary hypertension in patients with obstructive sleep apnea: the effect of continuous positive airway pressure on pulmonary hemodynamics Respiration. 2001. 68. P. 566–572.
4. Arias M.A., García–Río F., Alonso–Fernández A. et al. Pulmonary hypertension in obstructive sleep apnoea: effects of continuous positive airway pressure. European Heart Journal. 2006. 27. P. 1106–1113.
5. Arzt M., Luigart R., Schum C. et al. Sleep–disordered breathing in deep vein thrombosis and acute pulmonary embolism. Eur. Respir. J. 2012. 40. P. 919–24.
6. Bady E., Achkar A., Pascal S. et al. Daytime pulmonary hemodynamics in patients with obstructive sleep apnea without lung disease. Am. J. Respir. Crit. Care Med. 1999. 159. P. 1518–1526.
7. Bady E., Achkar A., Pascal S. et al. Pulmonary arterial hypertension in patients with sleep apnoea syndrome. Thorax. 2000. 55. P. 934–939.
8. Baessler A., Nadeem R., Harvey M. et al. Treatment for sleep apnea by continuous positive airway pressure improves levels of inflammatory markers — a meta–analysis. J. Inflamm.(Lond). 2013. 10:13. http://www.journal–inflammation.com/content/10/1/13. doi:10.1186/1476–9255–10–13
9. Bartsch P., Maggiorini M., Ritter M. et al. Prevention of high–altitude pulmonary edema by nifedipine. N. Engl. J. Med. 1991. 325. P. 1284–1289.
10. Berman E.J., DiBenedetto R.J., Causey D.E. et al. Right ventricular hypertrophy detected by echocardiography in patients with newly diagnosed obstructive sleep apnea. Chest. 1991. 100(2). P. 347–350.
11. Bitter T., Faber L., Hering D. et al. Sleep–disordered breathing in heart failure with normal left ventricular ejection fraction. Eur. J. Heart Fail. 2009. 11. P. 602–608.
12. Bonderman D., Jakowitsch J., Adlbrecht C. et al. Medical conditions increasing the risk of chronic thromboembolic pulmonary hypertension. Thromb. Haemost. 2005. 93. P. 512–516.
13. Brown L.M., Chen H., Halpern S. et al. Delay in recognition of pulmonary arterial hypertension: factors identified from the REVEAL. Registry Chest. 2011. 140. P. 19–26.
14. Chami H.A., Devereux R.B., Gottdiener J.S. et al. Left ventricular morphology and systolic function in sleep–disordered breathing: the Sleep Heart Health study Circulation. 2008. 117. P. 2599–2607.
15. Deflandre E. Degey S. Opsomer N. et al. Obstructive sleep apnea and smoking as a risk factor for venous thromboembolism events: review of the literature on the common pathophysiological mechanisms. Obes. Surg. 2016. 26. P. 640–648.
16. Friedman O., Bradley T.D., Chan C.T., Parkes R., Logan A.G. Relationship between overnight rostral fluid shift and obstructive sleep apnea in drug–resistant hypertension. Hypertension. 2010. 56. Р. 1077–1082.
17. Fuhrman C., Fleury B., Nguyên X.L., Delmas M.C. Symptoms of sleep apnea syndrome: high prevalence and underdiagnosis in the French population. Sleep Med. 2012. Vol. 13. 7. P. 852–858.
18. Galiè N., Hoeper M.M., Humbert M. et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: The Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur. Heart J. 2009. 30. P. 2493–2537.
19. Galiè N., Humbert M., Vachiery J.L. et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur. Respir. J. 2015. 46. P. 903–975.
20. Gottlieb D.J., Yenokyan G., Newman A.B. et al. Prospective study of obstructive sleep apnea and incident coronary heart disease and heart failure: the Sleep Heart Health study. Circulation. 2010. 122. P. 352–360.
21. Guilleminault C., Motta J., Mihm F., Melvin K. Obstructive sleep apnea and cardiac index. Chest. 1986. 89(3). P. 331–334.
22. Hedner J., Ejnell H., Caidahl K. Left ventricular hypertrophy independent of hypertension in patients with obstructive sleep apnoea. J. Hypertens. 1990. 8. P. 941–946.
23. Heinzer R., Vat S., Marques–Vidal P. et al. Prevalence of sleep–disordered breathing in the general population: the HypnoLaus study. Lancet Respir. Med. 2015. Published Online February 12, 2015. http://dx.doi.org/10.1016/ S2213–2600(15)00043–0.
24. Hetzel M., Kochs M., Marx N. et al. Pulmonary hemodynamics in obstructive sleep apnea: frequency and causes of pulmonary hypertension. Lung. 2003. 181. P. 157–166.
25. Hoeper M.M., Bogaard H.J., Condliffe R. et al. Definitions and diagnosis of pulmonary hypertension. J. Am. Coll. Cardiol. 2013. 62(Suppl.). Р. 42–50.
26. Imran T.F., Ghazipura M., Liu S. et al. Effect of continuous positive airway pressure treatment on pulmonary artery pressure in patients with isolated obstructive sleep apnea: a meta–analysis. Heart Failure Rewiews September. 2016. 21(5). P. 591–598.
27. Ip M.S., Lam B., Chan L.Y. et al. Circulating nitric oxide is suppressed in obstructive sleep apnea and is reversed by nasal continuous positive airway pressure. Am. J. Respir. Crit. Care Med. 2000. 162. P. 2166–2171.
28. Jilwan F.N., Escourrou P., Garcia G. et al. High occurrence of hypoxemic sleep respiratory disorders in precapillary pulmonary hypertension and mechanisms. Chest. 2013. 143. P. 47–55.
29. Kauppert C.A., Dvorak I., Kollert F. et al. Pulmonary hypertension in obesity–hypoventilation syndrome. Respir. Med. 2013. 107. P. 2061–2070.
30. Klinger J.R., Elliott C.G., Levine D.J. et al. Therapy for pulmonary arterial hypertension in adults. Update of the CHEST Guideline and Expert Panel Report. CHEST. 2019. 155(3). P. 565–586.
31. Kohler M., Stradling J.R. Mechanisms of vascular damage in obstructive sleep apnea. Nat. Rev. Cardiol. 2010. 7. P. 677–685.
32. Lam C.S., Roger V.L., Rodeheffer R.J. et al. Pulmonary hypertension in heart failure with preserved ejection fraction: a community–based study. J. Am. Coll. Cardiol. 2009. 53. P. 1119–1126.
33. Launay J.M., Herve P., Peoch K. et al. Function of the serotonin 5–hydroxytryptamine 2B receptor in pulmonary hypertension. Nat. Med. 2002. 8. P. 1129–1135.
34. Li J.M., Shah A.M. Endothelial cell superoxide generation: regulation and relevance for cardiovascular pathophysiology. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004. 287. Р. 1014–1030.
35. Liak C., Fitzpatrick M. Coagulability in obstructive sleep apnea. Can. Respir. J. 2011. 18. P. 338–48.
36. Marvisi M., Vento M.G., Balzarini L. et al. Continuous positive airways pressure and uvulopalatopharyngoplasty improves pulmonary hypertension in patients with obstructive sleep apnoea. Lung. 2015. 193(2). P. 269–274.
37. McLaughlin V.V., Archer S.L., Badesch D.B. et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents. J. Am. Coll. Cardiol. 2009. 53. P. 1573–1619.
38. McNicholas W.T., Bonsignore M.R. Sleep apnoea as an independent risk factor for cardiovascular disease: current evidence, basic mechanisms and research priorities. European Respiratory Journal. 2007. 29(1). P. 156–178.
39. Mereles D., Ehlken N., Kreuscher S. et al. Exercise and respiratory training improve exercise capacity and quality of life in patients with severe chronic pulmonary hypertension. Circulation. 2006. 114. P. 1482–1489.
40. Minai O.A., Ricaurte B., Kaw R. et al. Frequency and impact of pulmonary hypertension in patients with obstructive sleep apnea syndrome. Am. J. Cardiol. 2009. 104. P. 1300–1306.
41. Minic M., Granton J.T., Ryan C.M. Sleep disordered breathing in group 1 pulmonary arterial hypertension. J. Clin. Sleep Med. 2014. 10. P. 277–283.
42. Nahmias J., Lao R., Karetzky M. Right ventricular dysfunction in obstructive sleep apnoea: reversal with nasal continuous positive airway pressure. Eur. Respir. J. 1996. 9. P. 945–951.
43. Phillips B.G., Narkiewicz K., Pesek C.A. et al. Effects of obstructive sleep apnea on endothelin–1 and blood pressure. J.
Hypertens. 1999. 17. P. 61–66.
44. Raskin J., Qua D., Marks T., Sulica R. A retrospective study on the effects of pulmonary rehabilitation in patients with pulmonary hypertension. Chronic Respir. Dis. 2014. 11. P. 153–162.
45. Ryan S., Taylor C.T., McNicholas W.T. Selective activation of inflammatory pathways by intermittent hypoxia in obstructive sleep apnea syndrome. Circulation. 2005. 112. P. 2660–2667.
46. Sajkov D., Wang T., Saunders N.A. et al. Daytime pulmonary hemodynamics in patients with obstructive sleep apnea without lung
disease. Am. J. Respir. Crit. Care Med. 1999. 159. P. 1518–1526.
47. Sajkov D., Wang T., Saunders N.A. et al. Continuous po–sitive airway pressure treatment improves pulmonary hemodyna–mics in patients with obstructive sleep apnea. Am. J. Respir. Crit. Care Med. 2002. 165. P. 152–158.
48. Singh M., Liao P., Kobah S. et al. Proportion of surgical patients with undiagnosed obstructive sleep apnoea. Br. J.
Anaesth. 2013. 110(4). P. 629–636.
49. Smith M.L., Niedermaier O.N.W., Hardy S.M. et al. Role of hypoxemia in sleep apnea–induced sympathoexcitation. Journal of the Autonomic Nervous System. 1996. 56(3). P. 184–190.
50. Somers V.K., Dyken M.E., Skinner J.L. Autonomic and hemodynamic responses and interactions during the Mueller maneuver in humans. Journal of the Autonomic Nervous System. 1993. 44(2–3). P. 253–259.
51. Somers V.K., Mark A.L., Zavala D.C., Abboud F.M. Contrasting effects of hypoxia and hypercapnia on ventilation and sympathetic activity in humans. Journal of Applied Physiology. 1989. 67(5). P. 2101–2106.
52. Szulcek R., Happe C.M., Rol N. et al. Delayed microvascular shear adaptation in pulmonary arterial hypertension. Role of platelet endothelial cell adhesion molecule–1 cleavage. Am. J. Respir. Crit. Care Med. 2016. 193. P. 1410–1420.
53. Tilkian A.G., Guilleminault C., Schroeder J.S. Hemodynamics in sleep induced apnea. Studies during wakefulness and sleep. Annals of Internal Medicine. 1976. 85(6). P. 714–719.
54. Ulrich S., Keusch S., Hildenbrand F.F. et al. Effect of nocturnal oxygen and acetazolamide on exercise performance in patients with pre–capillary pulmonary hypertension and sleep–disturbed breathing: randomized, double–blind, cross–over trial. Eur. Heart J. 2015. 36. P. 615–623.
55. Virolainen J., Ventila M., Turto H., Kupari M. Effect of negative intrathoracic pressure on left ventricular pressure dyna–mics and relaxation. Journal of Applied Physiology. 1995. 79(2). P. 455–460.
56. Wang B., Yan B., Song D., Ye X., Liu SF. Chronic intermittent hypoxia down–regulates endothelial nitric oxide synthase expression by an NF–κB–dependent mechanism. Sleep Med. 2012. 14. P. 165–171.
57. Wang Z., Li AY., Guo Q.H. et al. Effects of cyclic intermittent hypoxia on ET–1 responsiveness and endothelial dysfunction of pulmonary arteries in rats. PLoS One. 2013. 8. e. 58078.
58. White L.H., Bradley T.D. Role of nocturnal rostral fluid shift in the pathogenesis of obstructive and central sleep apnoea. J. Physiol. 2013. 591. P. 1179–93.
59. Xue Yu, Zhiwei Huang, Yi Zhang et al. Obstructive sleep apnea in patients with chronic thromboembolic pulmonary hypertension. J. Thorac. Dis. 2018. 10(10). P. 5804–5812.