Резюме
Актуальність. У дослідженні подано аналіз багаторічної праці, виконаної під керівництвом члена-кореспондента НАМНУ, професора В.М. Єльського на кафедрі патологічної фізіології Донецького національного медичного університету імені М. Горького, з експериментального відтворення та аналізу синдрому ендогенної інтоксикації (СЕІ) при черепно-мозковій травмі (ЧМТ). Мета дослідження: розробити концепцію стадійності та ключових механізмів розвитку СЕІ при ЧМТ. Матеріали та методи. Дослідження проведено на 555 білих безпородних щурах-самцях; ЧМТ відтворювали за методикою В.М. Єльського, С.В. Зябліцева (2005). У крові визначали динаміку 30 показників гормонів, метаболітів, імунологічних та інших біологічно активних речовин; 7 показників активності ферментів; додатково розраховували 5 індексів (загалом у дослідженні вивчено 42 показники). Порушення функції центральної нервової системи оцінювали за методикою оцінки неврологічного дефіциту у балах за показниками рівня свідомості, стану рефлекторної сфери, слуху, м’язового тонусу тулуба та кінцівок, реакції на світло і біль, дихання, руху та деяких поведінкових реакцій. Проводили дослідження морфологічних показників ураження головного мозку. Результати. Аналіз динаміки вивчених показників та їх співвідношень дозволив виділити чотири стадії розвитку СЕІ: реактивно-метаболічну, запально-токсичну, стадію системної ендогенної інтоксикації (остаточний розвиток СЕІ) і стадію розвитку поліорганної недостатності (ПОН). Реактивно-метаболічна стадія характеризувалася: гіперергічною реакцією нейроендокринних систем з катаболічною дією; гіперглікемією, гіперінсулінізмом та інсулінорезистентністю; гіперметаболізмом. Запально-токсична стадія відзначалася: вираженою гіперферментемією, що вказувала на ушкодження клітинних мембран і мембран лізосом; проявами ендогенної інтоксикації (накопичення в крові молекул середньої маси і продуктів ліпопероксидації); генералізованими запальними реакціями (збільшення вмісту в крові медіаторів запалення, зокрема IL-1β, TNF-α, IL-6). Стадія системної ендогенної інтоксикації виділялася: різким накопиченням у крові лактату, що свідчило про приєднання системної гіпоксії; різким вторинним приростом молекул середньої маси з накопиченням низькомолекулярних і ароматичних пептидів; різким вторинним приростом прозапальних цитокінів; активацією ММP-9 і TIMP-1; різким приростом нейрональних білків S100В і NSE, що вказувало на прогресування ураження головного мозку. Стадія розвитку ПОН проявлялася: накопиченням у крові сечовини і креатиніну, ушкодженням еритроцитів, міокарда; різким приростом NSE; появою в крові великих кількостей антифосфоліпідних антитіл, що відбивало приєднання автоімунних процесів. Висновки. Отримані дані дозволили розробити концепцію розвитку СЕІ при ЧМТ і визначити ключові ланки його патогенезу, що може бути використано для розробки відповідних заходів патогенетичного лікування.
Актуальность. В исследовании представлен анализ многолетней работы, выполненной под руководством члена-корреспондента НАМНУ, профессора В.Н. Ельского на кафедре патологической физиологии Донецкого национального медицинского университета имени М. Горького, по экспериментальному воспроизведению и анализу синдрома эндогенной интоксикации (СЭИ) при черепно-мозговой травме (ЧМТ). Цель исследования: разработать концепцию стадийности и ключевых механизмов развития СЭИ при ЧМТ. Материалы и методы. Исследование проведено на 555 белых беспородных крысах-самцах; ЧМТ воспроизводили по методике В.Н. Ельского, С.В. Зяблицева (2005). В крови определяли динамику 30 показателей гормонов, метаболитов, иммунологических и других биологически активных веществ; 7 показателей активности ферментов; дополнительно рассчитывали 5 индексов (всего в исследовании изучено 42 показателя). Нарушение функции центральной нервной системы оценивали по методике оценки неврологического дефицита в баллах по показателям уровня сознания, состояния рефлекторной сферы, слуха, мышечного тонуса туловища и конечностей, реакции на свет и боль, дыхания, движения и некоторых поведенческих реакций. Проводили также исследование морфологических показателей повреждения головного мозга. Результаты. Анализ изученных показателей и их соотношений позволил выделить четыре стадии развития СЭИ: реактивно-метаболическую, воспалительно-токсическую, стадию системной эндогенной интоксикации (окончательное развитие СЭИ) и стадию развития полиорганной недостаточности (ПОН). Реактивно-метаболическая стадия характеризовалась: гиперергической реакцией нейроэндокринных систем с катаболическим действием; гипергликемией, гиперинсулинизмом и инсулинорезистентностью; гиперметаболизмом. Воспалительно-токсическая стадия отличалась: выраженной гиперферментемией, указывающей на повреждение клеточных мембран и мембран лизосом; проявлениями эндогенной интоксикации (накопление в крови молекул средней массы и продуктов липопероксидации), генерализованными воспалительными реакциями (увеличение содержания в крови медиаторов воспаления, в том числе IL-1β, TNF-α, IL-6). Стадия системной эндогенной интоксикации выделялась: резким накоплением в крови лактата, что свидетельствовало о присоединении системной гипоксии; резким вторичным приростом молекул средней массы с накоплением низкомолекулярных и ароматических пептидов; резким вторичным приростом провоспалительных цитокинов; активацией ММP-9 и TIMP-1; резким приростом нейрональных белков S100В и NSE, что указывало на прогрессирование повреждения головного мозга. Стадия развития ПОН проявлялась: накоплением в крови мочевины и креатинина, повреждением эритроцитов, миокарда; резким приростом NSE; выявлением в крови большого количества антифосфолипидных антител, что отражало присоединение аутоиммунных процессов. Выводы. Полученные данные позволили разработать концепцию развития СЭИ при ЧМТ и определить ключевые звенья его патогенеза, что может быть использовано для разработки соответствующих мероприятий патогенетического лечения.
Background. This study deals with the results of the analysis of longstanding work carried out under the direction of the Corresponding Member of the National Academy of Medical Sciences of Ukraine, prof. V.М. Elskyy, Head of the Pathophysiology Department of M. Gorky Donetsk National Medical University, on experimental reproduction and analysis of endogenous intoxication syndrome (EIS) in the brain injury. This work aimed to develop a conception of staging and key mechanisms for the development of EIS associated with brain injury. Materials and methods. The study was conducted on 555 white, non-breeding male rats Wistar; experimental brain injury was reproduced by the method of V.M. Yelskyy, S.V. Ziablitsev (2005). In blood, the dynamics of 30 indicators of hormones, metabolites, immunological and other biologically active substances was determined; 7 indicators of activity of enzymes and additionally counted 5 indices (in general in this study 42 indicators were studied). Disorders of the central nervous system function were evaluated according to the method of assessing the neurological deficiency (scores) by the indicators of the level of consciousness, the state of the reflex field, hearing, muscle tone of the body and extremities, reaction to light and pain, respiration, movement and some behavioral reactions. The morphological parameters of brain damage were also determined. Results. The analysis of the studied parameters dynamics and their relationships allowed distinguishing four stages of EIS development: reactive-metabolic, inflammatory-toxic, the stage of systemic endogenous intoxication (the final development of EIS) and the stage of development of multiple organ failure (MOF). The reactive-metabolic stage was characterized by hyperergic reaction of neuroendocrine systems with catabolic action; hyperglycemia, hyperinsulinemia and insulin resistance; hypermetabolism. The severe hyperenzymemia, indicating damage to the cell membranes and lysosomal membranes, manifestations of endogenous intoxication (accumulation in the blood of molecules of medium mass and products of lipoperoxidation), generalized inflammatory reactions (increased inflammatory blood mediators, including IL-1β, TNF-α, IL-6) are typical for the inflammatory-toxic stage. The stage of systemic endogenous intoxication is characterized by a sharp accumulation of lactate in blood, indicating the systemic hypoxia inclusion; a sharp secondary increase in the molecules of medium mass with the accumulation of low molecular weight and aromatic peptides; a sharp secondary increase in proinflammatory cytokines; activation of MMP-9 and TIMP-1; a sharp increase in neuronal proteins S100B and NSE, indicating the progression of brain damage. Stage of MOF development is characterized by the accumulation in the blood of urea and creatinine, damage to red blood cells, myocardium; a sharp increase in NSE; the appearance in the blood of large quantities of antiphospholipid antibodies, which reflected the inclusion of autoimmune processes. Conclusions. The obtained data allowed developing a concept for the development of EIS in brain injury and to identify the key components of its pathogenesis, which can be used to develop appropriate measures of pathogenetic treatment.
Вступ
Актуальність роботи обумовлена високим рівнем травматизму здебільшого осіб молодого та середнього віку, а отже, найбільш працездатної частини населення, високим рівнем летальності, ускладнень і, як наслідок, інвалідизації постраждалих [1]. Зі зростанням кількості техногенних катастроф та масової загибелі людей питання про проведення патогенетичного лікування в достатньому обсязі все більше цікавить клініцистів, оскільки рівень загибелі постраждалих із тяжкими травматичними пошкодженнями залишається високим [2, 3]. Серед усіх причин травматичної хвороби (ТХ) черепно-мозкова травма (ЧМТ) є однією з головних причин високої летальності й інвалідизації населення [2], що в Україні в 2–3 рази вища за такі показники розвинутих країн. Щорічно у світі від ЧМТ гинуть 1,5 млн осіб, а 2,4 млн стають інвалідами. Частота ЧМТ у середньому становить 3–4 випадки на 1000 населення. При цьому тяжкі форми ЧМТ зустрічаються у більше ніж 40 % постраждалих, з яких від 30 до 50 % гинуть. Серед тих, хто вижив, повне функціональне відновлення центральної нервової системи (ЦНС) спостерігається рідко [1–3].
Летальність при ТХ у ранньому періоді (протягом двох тижнів після травми) обумовлена високим розвитком місцевих і системних циркуляторних, нейроендокринних, метаболічних та імунних уражень [4]. До первинних ушкоджень при ЧМТ відносять деструктивно-дегенеративні процеси [5], які репрезентовані травматичними некрозами і дистрофіями, що розвиваються в момент патогенного механічного впливу. Цей процес ускладнюється насамперед накопиченням токсинів, метаболічними зсувами та гальмуванням енергоутворюючих шляхів. Інтоксикація при ТХ має системний характер [6], однак не зовсім зрозуміло, які саме ланки, коли і в якій послідовності ушкоджуються. Отже, не ясно, які саме реакції є патологічними і визначальними для запуску патохімічних каскадів ендогенної інтоксикації. Це дає підстави для обґрунтування необхідності всебічного вивчення механізмів формування синдрому ендогенної інтоксикації (СЕІ) та стадійності його розвитку для формулювання нової концепції патогенетичної ролі ендогенної інтоксикації при ТХ головного мозку.
Матеріали та методи
В рамках даної роботи всі експериментальні дослідження проведено згідно з положеннями «Європейської конвенції про захист хребетних тварин, які використовуються для дослідних та інших наукових цілей» (Страсбург, 1985), Законом України «Про захист тварин від жорстокого поводження» № 3447-IV від 21.02.2006 р., наказом МОЗ України № 690 від 23.09.2009 р. Для проведення експерименту брали білих безпородних щурів-самців вагою від 190 до 200 г, віком 6 місяців. Усього в експериментах використано 555 тварин, з яких у контрольну групу увійшли 105. Тварин було розподілено на 5 серій. У першій серії, де було 50 тварин, зокрема 5 контрольних, вивчали летальність. В інших чотирьох серіях вивчали динаміку 30 показників вмісту у крові гормонів, метаболітів, імунологічних та інших біологічно активних речовин; 7 показників активності ферментів та додатково розраховували 5 індексів (загалом у дослідженні вивчено 42 показники, що стосуються розвитку СЕІ). Тварин виводили з експерименту у кількості 7–8 на кожний строк обстеження (3, 24, 48, 72 години та 5 діб після травми). Отже, у кожній серії було використано 35–40 тварин. Тварин контрольної групи виводили з експерименту у кількості 5 на кожний строк у кожній серії. Летальності серед тварин контрольної серії не було.
У піддослідних тварин моделювання ЧМТ проводили за методом [7] вільно падаючого вантажу. Енергія удару дорівнювала 0,516 Дж. Летальність становила 82 % протягом 5 діб спостереження, що відповідало показникам, притаманним середньотяжкій та тяжкій ЧМТ [8]. Порушення функції головного мозку оцінювали за методикою оцінки неврологічного дефіциту у балах згідно зі шкалою оцінки ступеня неврологічного дефіциту [7] за показниками рівня свідомості; стану рефлекторної сфери, зокрема ширини та реакції зіниць на світло, рогівкового рефлексу; слуху; м’язового тонусу тулуба та кінцівок; реакції на світло і біль; дихання; руху та деяких поведінкових реакцій. Проводили також дослідження морфологічних показників ураження головного мозку. При розкритті головного мозку у тварин візуально були констатовані піднадкісткові, субдуральні та епідуральні гематоми, крім того, гематоми на основі черепа — у зоні протиудару. Було виявлено ділянки розтрощення мозкової тканини та дедриту в зоні удару, а також набряк гіпофіза.
Таким чином, у тварин мала місце закрита ЧМТ за наявності шкірної гематоми й перелому кісток склепіння черепа без зсуву, середньотяжкого ступеня з наявністю розтрощення кори тім’яних і скроневих часток (у зоні удару) й основи лобових і скроневих часток (у зоні протиудару); ушкодження речовини головного мозку у вигляді дифузійних дрібноточкових крововиливів; оболонкової гематоми — у зоні удару.
Для статистичної обробки отриманих результатів використовували програму Statistica 10 (StatSoft, Inc., США).
Результати та обговорення
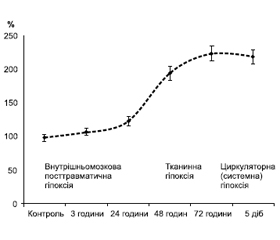
СЕІ при ЧМТ формується внаслідок розгортання цілої низки патологічних процесів, які запускаються під впливом травмуючого агента, охоплюють головний мозок, а потім усі органи та системи організму [9–12]. Підґрунтям цього стану, на нашу думку, є поетапне розгортання патологічних реакцій у різних системах регуляції гомеостазу, комплексне вивчення яких в умовах експерименту дозволило виділити чотири стадії розвитку СЕІ (рис. 1).
Безпосередньо після травматичного впливу розвинулася реактивно-метаболічна стадія. Потім, починаючи з 24-ї години після травми, на перший план виходили запальні процеси та накопичення токсинів (запально-токсична стадія). Починаючи з третьої доби після травми (після 48 годин) формувалася стадія системної ендогенної інтоксикації, що знаменувала собою остаточний розвиток СЕІ, а з четвертої доби, як наслідок системної дії токсинів, формувалася поліорганна недостатність (ПОН).
На рис. 2 подано основні показники, що характеризували розвиток СЕІ при ЧМТ. Початок реактивно-метаболічної стадії ініціювався функціональною активацією центральних ланок нейроендокринної системи за гіперергічним типом. Це проявлялося з перших годин після травми і тривало до 72 годин. Приріст рівня кортикостероїду (КС), основного глюкокортикоїдного гормона, був зумовлений активуючим впливом АКТГ. Рівень ВП у цей період також був трикратно підвищений, а наявність позитивного зв’язку АКТГ і ВП указувала на співдружність цієї реакції, викликаної, очевидно, відповідними нейрохімічними механізмами, що спричиняли активацію нейроендокринних систем з катаболічною дією.
/89.jpg)
Глюкокортикоїди активують глюконеогенез у печінці за рахунок впливу на транскрипцію відповідних ферментів; збільшують вміст у крові субстратів глюконеогенезу (амінокислот) через посилення протеолізу у м’язах; зменшують поглинання глюкози глюкозозалежними тканинами [4]. Результатом цього каскаду різноманітних біохімічних реакцій є загальна активація метаболізму і, зокрема, гіперглікемія [13]. Вміст глюкози в крові в реактивно-метаболічну стадію виявився істотно і статистично значуще підвищеним у всі терміни спостереження, особливо через 3 і 24 години після травми. Активація утворення глюкози й інших легко засвоюваних джерел енергії (амінокислоти, тригліцериди, жирні кислоти), на нашу думку, була потужною захисною реакцією ЦНС на травму.
Гіперглікемія супроводжувалася різко вираженим збільшенням вмісту в крові рівня інсуліну (ІН) — гіперінсулінізмом [9]. Оцінка динаміки індексу HOMA-ІР виявила десятикратний приріст його величини вже через 3 години після травми, що довело наявність інсулінорезистентності, яка могла розвиватися внаслідок реалізації ефектів катехоламінів і глюкокортикоїдів на інсулінорезистентні тканини (інгібування фосфорилювання глюкози в гексокіназній реакції) [13]. Останнє було підтверджено виявленим зниженням чутливості тканин до ІН (за результатами аналізу динаміки індексу чутливості до інсуліну — QUICKI).
Надалі динаміка аналізованих показників дозволила виявити типові патологічні процеси — запалення й інтоксикацію (рис. 2), що починалися вже в попередню стадію, оскільки одні патологічні процеси запускають динамічний розвиток інших, як-от: гіперергічна реакція нейроендокринних систем → гіперметаболізм → нагромадження токсинів → запуск запальних реакцій → поглиблення ендотоксемії [10, 14].
Насамперед необхідно зазначити, що, починаючи з цього періоду, формувалася виражена гіперферментемія [11, 15, 16]. Багаторазово підвищувалася активність ферментів, що відбивають клітинну деструкцію (амінотрансферази), ушкодження клітинних мембран (ЛФ), лабілізацію й ушкодження лізосомальних мембран (КФ і КD). Останнє підтверджувало розвиток запальної реакції, оскільки саме лізосомальні ферменти є маркерами альтеративної стадії запалення. Оскільки амінотрансферази й ЛФ є маркерами процесів ушкодження клітин — цитолізу, можна було стверджувати, що виявлена гіперферментемія в ранній термін ТХ головного мозку була відбиттям посттравматичної деструкції тканини головного мозку, а в пізні — відображала залучення до патологічного процесу інших органів і тканин організму (розвиток ПОН).
Основним проявом другої стадії ТХ головного мозку було накопичення в крові молекул середньої маси (МСМ) [14], що мало трифазовий перебіг, тобто розвиток ендотоксикозу проходив три етапи (рис. 3).
/90.jpg)
Первинне накопичення (3 години після травми), далі — період типу «плато», коли рівень МСМ істотно не змінювався (24–48 годин після травми) і, нарешті, лавиноподібне накопичення МСМ, починаючи з 3-ї доби після травми (72 години — 5 діб). Перший етап був пов’язаний із надходженням у кров ендотоксинів з ушкодженої тканини головного мозку (первинний — «мозковий» посттравматичний ендотоксикоз). На цьому етапі переважали нуклеотиди й неароматичні амінокислоти. Причиною наступної стабілізації накопичення токсинів у крові могло бути їх руйнування та виведення нирками (період «плато»). Третій етап починався з третьої доби після травми та характеризувався вираженим приростом усіх фракцій МСМ, що було пов’язано, на наш погляд, не тільки з прогресуванням ураження нервової тканини, але й із системним ушкодженням внутрішніх органів і тканин організму внаслідок остаточного формування на цьому етапі СЕІ та розвитку ПОН. Крім нуклеотидів і неароматичних амінокислот, на цьому етапі значно збільшувалася кількість низькомолекулярних пептидів і ароматичних амінокислот, що було несприятливим фактором.
Істотним показником розвитку СЕІ у другу стадію було також нагромадження в крові продуктів ПОЛ — ДК і МДА [14]. У кількісному відношенні накопичення в крові обох продуктів ПОЛ відбувалося паралельно і приблизно рівною мірою. Фаза первинного накопичення (результат процесів посттравматичної нейродеструкції) змінювалася фазою «плато» через 48 годин після травми. Починаючи з цього терміну середні величини, що відбивали вміст як ДК, так і МДА, статистично не відрізнялися між собою.
У відповідь на ушкоджувальну дію ендотоксинів у другу фазу СЕІ формувалася запальна реакція [10, 11, 14]. Показниками цього процесу були збільшення вмісту в крові циркулюючих імунних комплексів (ЦІК), білків гострої фази запалення (С-реактивного білка (СРБ) і церулоплазміну) й активація інтерлейкінового каскаду. Багаторазове підвищення рівня в крові ЦІК, починаючи з 24-ї годин після травми, дозволило вважати цей термін початком генералізованої запальної реакції. Це, у свою чергу, могло спричинити автоалергічне ушкодження базальної мембрани ниркових клубочків і підендотеліального шару капілярів та, відповідно, формування ниркової недостатності й ендотеліальної дисфункції.
Динаміка вмісту в крові СРБ також підтверджувала наявність генералізованої запальної реакції — його рівень через 24 години після травми перевищував конт-рольний у 10,5 раза. Прогресуючий приріст СРБ надалі вказував на прогресування та генералізацію запальної реакції.
Динаміка церулоплазміну, що належить до білків гострої фази запалення, була двофазною — збільшення в першу добу після травми з подальшим наростаючим зниженням, що вказувало на прогресування патологічних процесів, починаючи з другої доби після травми.
Отже, протягом першої-другої доби після травми запускалися запально-токсичні процеси, що знайшло своє відбиття в прогресуючому накопиченні в крові ЦІК, МСМ, продуктів ПОЛ, токсичних метаболітів, а також в активації синтезу білків гострої фази — СРБ і церулоплазміну.
Також у цей період активувався цитокіновий каскад, спостерігалася так звана цитокінова буря [14]. Загальною закономірністю реакції цитокінового каскаду став максимальний приріст вивчених інтерлейкінів (IL-1β, TNF-a, IL-6, IL-8) у 1-шу — 2-гу добу після травми, що відбивало активацію, а згодом — генералізацію запальної реакції. Первинний, відносно помірний приріст цитокінів був реакцією імунної системи у відповідь на ушкодження і сприяв мобілізації захисних сил організму, тоді як наступне наростання знаменувало початок ушкодження та формування запальної стадії перебігу ТХ головного мозку. Основним реактантом імунної системи при ЧМТ був IL-1β [11]. Далі (у міру зменшення ступеня реагування) розташувалися IL-6 і TNF-a, тоді як рівень IL-8 навіть знижувався через 72 години — 5 діб після травми. Вторинний різкий приріст вмісту в крові IL-1β при стабілізації активності первинних медіаторів запалення (КФ і КD) на рівні 24 годин указував на те, що в цей період запальний процес набував якісно нової властивості — відбувалася його генералізація із залученням інших органів і систем.
Наступною стадією розвитку СЕІ при ТХ головного мозку була стадія системної ендогенної інтоксикації, що формувалася на другу добу перебігу посттравматичного періоду (48 годин після травми). В цей період у результаті метаболічних зрушень, накопичення в крові токсинів, генералізації запальної реакції і прогресування гіпоксії патологічні зрушення набували системного характеру [9, 12]. Пусковою ланкою цієї реакції була системна ендогенна інтоксикація, а СЕІ сягав максимального розвитку (рис. 2). У нашому дослідженні наявність гіперметаболізму поряд з гіперглікемією та динамікою ПМ підтверджувала накопичення в крові лактату, що відбивало порушення гліколітичних процесів і утворення АТФ. Це проявлялося, починаючи з 48-ї години після травми, і знаменувало собою формування ушкоджень гіпоксичного генезу. Саме накопичення в крові лактату дозволило констатувати розвиток при ЧМТ системної гіпоксії, що була наслідком патологічних процесів, які розгортаються в реактивно-метаболічну стадію. Гіпоксія при ЧМТ має змішаний характер і розвивається внаслідок дії токсинів на тканини (тканинна гіпоксія), а згодом — у результаті порушення мікроциркуляції й зниження продуктивності серця (циркуляторна гіпоксія). З огляду на прогресування накопичення в крові лактату можна вважати, що гіпоксія наростала та була прогресуючим патологічним процесом, що мав постійний перебіг і на фоні якого поетапно розвивався СЕІ. Динаміку накопичення в крові лактату і схематичне зображення етапів розвитку гіпоксії при ЧМТ наведено на рис. 4.
/91.jpg)
Порівнюючи дані рис. 4 з розвитком стадій СЕІ (рис. 1), можна стверджувати, що гіпоксія починається в запально-токсичній стадії, підсилюється при формуванні стадії системної ендогенної інтоксикації і досягає максимального вираження в стадію формування ПОН. У цю стадію було відзначене також і виснаження АОС, яке проявлялося зниженням активності СОД, що досягло катастрофічно низького рівня через 5 діб після травми, коли активність ферменту становила всього 6 % від контрольного рівня [12]. Активність каталази, як і СОД, прогресивно знижувалася, що досягало статистично значущого рівня, починаючи з 48-ї години після травми. Активація матриксних металопротеїназ відбувається при запаленні й визначає запуск проліферативної стадії, а їх здатність руйнувати рецептори клітинних мембран і сприяти викиду медіаторів апоптозу, FAS-ліганду дозволяє вважати їх важливими регуляторними факторами. Вміст ММР-9 збільшувався тільки з третьої стадії перебігу ТХ головного мозку і надалі прогресивно наростав [14], що вказувало, на наш погляд, на активацію процесів розпаду тканини, які починалися стрімко, про що свідчило багаторазове збільшення ММР-9. Вміст TIMP-1 також мав тенденцію до підвищення, однак статистично значущим приріст цього показника був тільки через 72 години і 5 діб після травми. Отже, різке збільшення вмісту ММР-9 і TIMP-1 вказувало на подальший розвиток запального процесу та, як і динаміка вивчених прозапальних інтерлейкінів, підтверджувало генералізацію запальної реакції при ЧМТ [11]. ММР-9 і TIMP-1 указували на прогресування розладів гомеостазу, а різкий приріст їх вмісту у крові через 48 і 72 години свідчив про розвиток СЕІ.
Важливою особливістю стадії системної ендогенної інтоксикації було прогресуюче накопичення в крові маркерів ураження головного мозку — нейрональних білків (S100В і NSE) [15]. Крім власне деструкції нейронів, підвищення їх вмісту у крові відбивало підвищення проникності гематоенцефалічного бар’єра. Зростання вмісту нейрональних білків у крові є пре-диктором летального кінця при тяжкій ЧМТ і розвитку посттравматичної енцефалопатії при середньотяжкій і легкій ЧМТ [16].
Рівень у крові білка S100B через 48 годин після травми був статистично вірогідно вищим, ніж у попередньому періоді [15]. Це вказувало на те, що приріст відбувався не поступово, а стрибкоподібно з двома піками: перший — через 3 години, а другий — через 48 годин після травми. Очевидно, у цьому періоді відбувалося посилення деструкції нервової тканини внаслідок розвитку тканинної гіпоксії, збільшення енергодефіциту і поглиблення розвитку системної токсемії — СЕІ. Як і S100B, NSE характеризував ступінь гіпоксичного й постішемічного ураження головного мозку [16]. Характер приросту рівня NSE був поступовим, з деяким приростом через 48 годин після травми, коли він значуще перевищив такий після 24 годин. Цей факт, як і у випадку з білком S100B, засвідчував різке погіршення стану мозкової тканини на другу добу після травми.
Необхідно зважати на те, що білок S100В при високій концентрації підсилює нейродегенерацію шляхом запуску S100В-індукованого апоптозу, а в мікромолярних концентраціях індукує і некроз [16, 17]. Цей ефект реалізується через посилення експресії в нервовій тканині IL-1b і IL-6, а через активацію утворення оксиду азоту — також і IL-8 і TNF-α. У свою чергу, ці інтерлейкіни індукують експресію S100B. Отже, замикається ще одне патогенетичне коло нейронального ушкодження.
На наступній, заключній, стадії в результаті розгортання СЕІ формувалася ПОН (рис. 2). Свідченням порушення функції печінки й нирок було накопичення в крові сечовини і креатиніну. Дворазове їх наростання в крові через 72 години і 5 діб після травми відбивало декомпенсацію метаболічних порушень цього періоду, що стало наслідком системної патогенної дії ендогенних токсинів.
Про постійне наростання ушкодження еритроцитів свідчив аналіз динаміки простагландину Е (ПГЕ), що у цю стадію перевищував контрольний рівень у 2,8–3,0 раза [12]. Через 72 години після травми зафіксовано вторинний, уже трикратний, приріст показника. Така динаміка була одним із проявів ендотоксикозу при СЕІ. Крім того, ушкодження еритроцитів сигналізувало про приєднання кров’яної (гемічної) гіпоксії внаслідок зниження кількості нормально функціонуючих еритроцитів і, відповідно, кисневої ємності крові.
Через 72 години і 5 діб після травми рівні нейрональних білків істотно перевищували вихідні значення, що доводило прогресуючий характер ушкодження нервової тканини та порушення функції гематоенцефалічного бар’єра в пізні терміни після травми [15].
Ще однією ланкою патогенезу розвитку СЕІ при ТХ головного мозку була виявлена автоімунна реакція — новоутворення АФЛ [18]. Накопичення антитіл у першу добу після травми можна було розцінювати як захисну реакцію, спрямовану на елімінацію з крові нейрональних антигенів. Значне ж їх накопичення у крові в більш пізній період указувало на гіпергічну автоімунну реакцію, що може спричиняти ушкодження тромбоцитів і ендотелію судин. Так, вміст АТ-КЛ стрімко збільшувався і через 5 діб після травми перевищував контрольні значення у 24,3 раза. Подібну, але менш виражену реакцію мала динаміка вмісту в крові АФЛ-IgМ і АФЛ-IgG. Це свідчило про те, що в цей період ТХ головного мозку поряд з генералізацією запалення запускаються також і автоімунні реакції.
Отже, показано, що в четверту стадію ТХ головного мозку відбувалося прогресування ураження головного мозку, внутрішніх органів, клітин крові. Крім того, було зафіксовано й ураження серця [19]. Так, наростання активності ЛДГ можна було вважати загальною неспецифічною реакцією, характерною для ПОН та ураження серця. Активність КК-МВ через 72 години і 5 діб після травми була підвищеною в 6,1–6,6 раза. Системність клітинного ушкодження підтверджувало різке підвищення активності трансаміназ (АлАТ і АсАТ) з переважним збільшенням активності кардіоспецифічного ферменту — АсАТ. Безпосереднім підтвердженням розвитку ушкодження міокарда на 5-ту добу після травми було збільшення активності КК-МВ, що істотно на цей термін випереджало приріст активності трансаміназ. Нарешті, доводило розвиток гіпоксичного ураження серця різке (більше ніж тридцятикратне) збільшення вмісту тропоніну I. На нашу думку, причина ушкодження міокарда при ТХ головного мозку за типом посттравматичної міокардіодистрофії полягала в патологічній дії ендогенних токсинів, які в цей період у надлишкових кількостях накопичувалися в організмі.
Висновки
Проведене дослідження дозволило виділити чотири стадії розвитку СЕІ: реактивно-метаболічну, запально-токсичну, стадію системної ендогенної інтоксикації (остаточний розвиток СЕІ) і стадію розвитку ПОН. Наведено їх часову динаміку, описано типові патологічні процеси та прояви.
Конфлікт інтересів. Автори заявляють про відсутність конфлікту інтересів і власної фінансової зацікавленості при підготовці даної статті.
Список литературы
1. Ельский В.Н. Патофизиология, диагностика и интенсивная терапия тяжелой черепно-мозговой травмы. Донецк: Новый мир, 2004. 200 с.
2. Хобзей Н.К. Эпидемиология инвалидности вследствие черепно-мозговых травм в Украине. Здоров’я нації. 2011. № 3. С. 30-34.
3. Pineda J.A. Mortality in severe traumatic brain injury — Authors’ reply. Lancet Neurol. 2013. Vol. 12, № 5. P. 427-428.
4. Ельский В.Н. Нейрогормональные регуляторные механизмы при черепно-мозговой травме. Донецк: Новый мир, 2008. 240 с.
5. Молдованов М.А. Диагностика и мониторинг нейронального повреждения при тяжелой черепно-мозговой травме. Общая реаниматология. 2010. Т. 1, № 1. С. 17-21.
6. Усенко Л.В. Патобиохимические особенности головного мозга при критических состояниях организма и обоснование метаботропной терапии: ч. 1. Некоторые особенности метаболизма головного мозга в физиологических условиях. Український журнал екстремальної медицини імені Г.О. Можаєва. 2009. Т. 10, № 1. С. 12-20.
7. Ельский В.Н. Моделирование черепно-мозговой травмы. Донецк: Новый мир, 2008. 140 с.
8. Nayak C. Relationship between markers of lipid peroxidation, thiol oxidation and Glasgow coma scale scores of moderate head injury patients in the 7 days post-traumatic period. Neurol. Res. 2008. Vol. 30, № 5. P. 461-464.
9. Зяблицев С.В. Механизмы и ключевые звенья развития синдрома эндогенной интоксикации при черепно-мозговой травме. Міжнародний вісник медицини. 2013. Т. 6, № 1. С. 18-23.
10. Ельский В.Н. Роль нейроиммуноэндокринных механизмов в формировании синдрома эндогенной интоксикации при травматической болезни. Таврический медико-биологический вестник. 2012. Т. 15, № 3. Ч. 1(59). С. 115-117.
11. Зяблицев С.В. Системные проявления неспецифической воспалительной реакции при травматической болезни головного мозга. Травма. 2012. Т. 13, № 4. С. 85-88.
12. Зяблицев С.В. Состояние реактивности нейрогормональных систем при травматической болезни головного мозга. Клінічна та експериментальна патологія. 2012. Т. XI, № 3(41). Ч. 1. С. 83-86.
13. Ziablitsev S.V. Disorders of carbohydrate metabolism in experimental brain injury. Фізіологічний журнал. 2016. Т. 62, № 4. С. 18-22.
14. Ziablitsev S.V. Systemic effects of unspecific inflammatory reaction at traumatic brain injury. Фізіологічний журнал. 2016. Т. 62, № 1. С. 67-72.
15. Зябліцев С.В. Динаміка вмісту нейроспецифічних білків та їх утворення при експериментальній черепно-мозковій травмі. Патологія. Науково-практичний медичний журнал. 2016. № 1(36). С. 49-53.
16. Lam A.G.M. Mechanism of glial activation by S100B: involvement of the transcription factor NFκB. Neurobiology of Aging. 2001. Vol. 22. P. 765-772.
17. Гудима А.А. Роль апоптозу в системних проявах тяжкої травми. Тернопіль: Укрмедкнига, 2010. 241 с.
18. Зяблицев С.В. Динаміка маркерів посттравматичного та аутоімунного пошкодження при черепно-мозковій травмі. Нейронауки: теоретичні та клінічні аспекти. 2013. Т. 9., № 1–2. С. 17-20.
19. Зябліцев С.В. Механізми формування посттравматичної міокардіодистрофії при черепно-мозковій травмі. Таврический медико-биологический вестник. 2012. Т. 15, № 4(60). С. 403-406.