
Международный эндокринологический журнал Том 17, №1, 2021
Вернуться к номеру
Сучасні погляди на генетичну детермінованість СТГ-секретуючих аденом гіпофіза (огляд літератури та власні дослідження)
Авторы: Ніколаєв Р.С.(1), Rostomyan L.(2), Beckers A.(2), Хижняк О.О.(1, 3), Микитюк М.Р.(1, 3), Караченцев Ю.І.(1, 3), Хазієв В.В.(1)
(1) — ДУ «Інститут проблем ендокринної патології ім. В.Я. Данилевського НАМН України», м. Харків, Україна
(2) — Centre Hospitalier Universitaire de Liège, Liège Université, Liège, Belgium
(3) — Харківська медична академія післядипломної освіти МОЗ України, м. Харків, Україна
Рубрики: Эндокринология
Разделы: Клинические исследования
Версия для печати
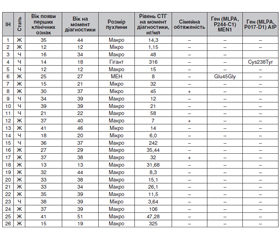
Актуальність. В роботі наведений огляд сучасних даних літератури щодо генетичної складової в етіології та патогенезі гормонально активної аденоми гіпофіза, що секретує соматотропний гормон (СТГ) і клінічними проявами якої є синдром акромегалії та/або гігантизму — синдром множинної ендокринної неоплазії 1, синдром Мак-К’юна — Олбрайта, комплекс Карні, акрогігантизм (Х-зчеплений), сімейні ізольовані аденоми гіпофіза (FIPA). Матеріали та методи. Для виявлення мутацій в гені AIP та з метою верифікації FIPA були обстежені 26 хворих української популяції (19 жінок та 7 чоловіків), в яких акромегалія була діагностована в підлітковому або молодому віці, і проведений генетичний аналіз. Для визначення генетичної детермінованості щодо розвитку СТГ-секретуючої аденоми гіпофіза та диференціальної діагностики синдромів FIPA та MEN1 методом секвенування (MLPA — Ligation-dependent Probe Amplification) було проведене дослідження генів (MLPA, P244-C1) за участю екзонів 1–6 MEN1, (MLPA, P017-D1) AIP. Результати. Серед обстежених тільки у двох осіб були визначені мутації гена AIP. В одного хворого генетичний скринінг на мутацію гена MEN1 був негативним, і жодних клінічних симптомів, що свідчать про синдром Мак-К’юна — Олбрайта, не було виявлено. Варіант гетерозиготного місенсу c.714C>G (p.Cys238Trp) виявлений у гені AIP. Цей аналіз гена AIP сумісний з генетичною схильністю до аденом гіпофіза. У нащадків даного пацієнта 50 % шансів успадкувати цей варіант. У іншої хворої з діагнозом «синдром множинної ендокринної неоплазії типу 1 (синдром Вермера): інсулінома, аденоми прищитоподібних залоз (2), первинний гіперпаратиреоз» встановлений варіант гетерозиготного місенсу c.134A>G (p.Glu45Gly), що був виявлений у гені MEN1. Варіант c.l34A>G (p.Glu45Gly), клас 4, ймовірно, є патогенним. Поширеність цього варіанта в загальній популяції невідома, тому він є дуже рідкісним. Висновки. Проведення генетичного аналізу є доцільним у хворих дитячого та молодого віку або в осіб, у яких СТГ-секретуюча макро-/гігантська аденома гіпофіза була діагностована в молодому віці (до 35 років), незалежно від сімейної обтяженості щодо аденом гіпофіза. У хворих з обтяженою спадковістю генетичний аналіз доцільно проводити у будь-якому разі для виявлення FIPA та прогнозування подальшого перебігу захворювання та ефективності лікування аналогами соматостатину.
Актуальность. В работе представлен обзор современных данных литературы о роли генетической составляющей в этиологии и патогенезе гормонально активной аденомы гипофиза, секретирующей соматотропный гормон (СТГ), клиническими проявлениями которой являются синдром акромегалии и/или гигантизма — синдром множественной эндокринной неоплазии 1 (МЭН-1), синдром Мак-Кьюна — Олбрайта, комплекс Карни, акрогигантизм (Х-сцепленный), семейная изолированная аденома гипофиза (FIPA). Материалы и методы. Для выявления мутаций в гене AIP и с целью верификации FIPA были обследованы 26 больных украинской популяции (19 женщин и 7 мужчин), у которых акромегалия была диагностирована в подростковом или молодом возрасте, и проведен генетический анализ. Для определения генетической детерминированности в развитии СТГ-секретирующей аденомы гипофиза и дифференциальной диагностики синдромов FIPA и МЭН-1 методом секвенирования (MLPA — Ligation-dependent Probe Amplification) было проведено исследование генов (MLPA, P244-C1) с участием экзонов 1–6 MEN1, (MLPA, P017-D1) AIP. Результаты. Среди обследованных только у двоих больных были выявлены мутации гена AIP. У одного больного генетический скрининг на мутацию гена MEN1 был отрицательным, и никаких клинических симптомов, свидетельствующих о синдроме Мак-Кьюна — Олбрайта, не было выявлено. Вариант гетерозиготного миссенса c.714C>G (p.Cys238Trp) обнаружен в гене AIP. Этот анализ гена AIP совместим с генетической предрасположенностью к развитию аденомы гипофиза. У потомков данного пациента 50 % шансов унаследовать этот вариант. У другой больной с диагнозом «синдром МЭН типа 1 (синдром Вермера): инсулинома, аденомы паращитовидных желез (2), первичный гиперпаратиреоз» установлен вариант гетерозиготного миссенса c.134A>G (p.Glu45Gly), который был выявлен в гене MEN1. Вариант c.l34A>G (p.Glu45Gly), класс 4, вероятно, патогенный. Распространенность этого варианта в общей популяции неизвестна, поэтому он очень редок. Выводы. Проведение генетического анализа целесообразно у больных детского и молодого возраста или у лиц, у которых СТГ-секретирующая макро-/гигантская аденома гипофиза была диагностирована в молодом возрасте (до 35 лет) независимо от семейного анамнеза. У больных с отягощенной наследственностью генетический анализ целесообразно проводить в любом случае для выявления FIPA и прогнозирования дальнейшего течения заболевания и эффективности лечения аналогами соматостатина.
Background. This article presents a review of the current literature on the role of the genetic component in the etiology and pathogenesis of hormone-active pituitary adenomas secreting growth hormone (GH) and clinically manifesting by acromegaly and/or gigantism (multiple endocrine neoplasia 1 (MEN-1), McCune-Albright syndrome, Carney complex, X-linked acrogigantism (X-LAG), familial isolated pituitary adenoma — FIPA). Materials and methods. To identify mutations in the AIP gene and to verify FIPA, 26 patients of the Ukrainian population (19 women and 7 men) were examined in whom acromegaly was diagnosed in adolescence or young age, and genetic analysis was performed. To determine the genetic determinism in the development of GH-secreting pituitary adenoma and differential diagnosis of FIPA and MEN-1 syndromes by sequencing method (MLPA — ligation-dependent probe amplification), the genes MLPA, P244-C1 were studied involving exons 1–6 MEN1 gene, (MLPA, P017-D1) AIP gene. Results. Among those examined, only two patients had AIP gene mutations. In one patient, genetic screening for MEN1 gene mutation was negative and no clinical symptoms suggestive of McCune-Albright syndrome were detected. A variant heterozygous missense c.714C>G (p.Cys238Trp) was found in the AIP gene. This AIP gene assay is compatible with a genetic predisposition to develop pituitary adenoma. The offspring of this patient has a 50% chance of inheriting this variant, acromegaly, hypersomatotropinemia, MEN-1 syndrome, familial isolated pituitary adenoma. Another patient was diagnosed with MEN syndrome type 1 (Wermer syndrome): insulinoma, parathyroid gland adenomas (2), primary hyperparathyroidism with a heterozygous c.134A>G variant (p.Glu45Gly) found in the MEN1 gene. The presence of the c.l34A>G (p.Glu45Gly) class variant 4 is likely to be pathogenic. The prevalence of this variant in the general population is unknown, so it is very rare. Conclusions. The genetic analysis is appropriate in pediatric and young patients or those with GH-secreting macro/giant pituitary adenoma diagnosed at a young age (under 35), regardless of family history. In patients with a history of a disease, genetic analysis is recommended in any case to identify FIPA and to predict the further course of the disease and the effectiveness of treatment with somatostatin analogues.
акромегалія; гіперсоматотропінемія; синдром МЕН-1; сімейна ізольована аденома гіпофіза; генетичний аналіз
акромегалия; гиперсоматотропинемия; синдром МЭН-1; семейная изолированная аденома гипофиза; генетический анализ
acromegaly; hypersomatotropinemia; MEN-1 syndrome; familial isolated pituitary adenoma; genetic analysis
Вступ
/21.jpg)
/22.jpg)
Матеріали та методи
Результати
/23.jpg)
Обговорення
Висновки
- Lavrentaki A., Paluzzi A., Wass J.A., Karavitaki N. Epidemiology of acromegaly: review of population studies. Pituitary. 2017. 20(1). 4-9. doi: 10.1007/s11102-016-0754-x.
- Maione L., Chanson P. National acromegaly registries. Best Pract. Res. Clin. Endocrinol. Metab. 2019. 33(2). 101264. doi: 10.1016/j.beem.2019.02.001.
- Khyzhnyak O., Mikityuk M., Guk M., Nikolaiev R., Gogitidze T. Clinical and hormonal features of acromegaly in patients from a Ukrainian neuroendocrinology centre. Probl. Endocr. Pathol. 2019. 2. 119-130. doi: 10.21856/j-PEP.2019.2.17.
- Gao M., Zhu B., Xu Z., Liu S., Liu J., Zhang G., Gao Y. et al. Association between acromegaly and a single nucleotide polymorphism (rs2854744) in the IGFBP3 gene. BMC Med. Genet. 2018. 19(1). 182. doi: 10.1186/s12881-018-0698-2.
- Sapochnik M., Nieto L.E., Fuertes M., Arzt E. Molecular mechanisms underlying pituitary pathogenesis. Biochem. Genet. 2016. 54(2). 107-19. doi: 10.1007/s10528-015-9709-6.
- Rostomyan L., Daly A.F., Petrossians P., Nachev E., Lila A.R., Lecoq A., Lecumberri B. et al. Clinical and genetic characterization of pituitary gigantism: an international collaborative study in 208 patients. Endocr. Relat. Cancer. 2015. 22(5). 745-57. doi: 10.1530/ERC-15-0320.
- Nikolaiev R., Standel S., Khyzhnyak O., Mikityuk M., Manska K. Features of hereditary aptitude to the development of the pituitary adenoma according to the data of the Ukrainian neuro-endocrinological center. Probl. Endocr. Pathol. 2020. 3. 71-80. doi: 10.21856/j-PEP.2020.3.09.
- Iacovazzo D., Hernández-Ramírez L.C., Korbonits M. Sporadic pituitary adenomas: the role of germline mutations and recommendations for genetic screening. Expert Rev. Endocrinol. Metab. 2017. 12(2). 143-153. doi: 10.1080/17446651.2017.1306439.
- Hernández-Ramírez L.C., Korbonits M. Familiar pituitary adenomas. Pituitary Disorders: Diagnosis and Management. 1st ed. John Wiley & Sons. 2013. 87-110.
- Verges B., Boureille F., Goudet P., Murat A. Pituitary disease in MEN type 1 (MEN1): data from the France-Belgium MEN1 multicenter study. J. Clin. Endocrinol. Metab. 2002. 87(2). 457-65. doi: 10.1210/jc.87.2.457.
- Horvath A., Stratakis C.A. Clinical and molecular genetics of acromegaly: MEN1, Carney complex, McCune-Albright syndrome, familial acromegaly and genetic defects in sporadic tumors. Rev. Endocr. Metab. Disord. 2008. 9(1). 1-11. doi: 10.1007/s11154-007-9066-9.
- Hernández-Ramírez L.C., Gabrovska P., Dénes J., Stals K., Trivellin G., Tilley D., Ferrau F. et al. Landscape of familial isolated and young-onset pituitary adenomas: prospective diagnosis in AIP mutation carriers. J. Clin. Endocrinol. Metab. 2015. 100(9). E1242-54. doi: 10.1210/jc.2015-1869.
- Daly A.F., Beckers A. Familial isolated pituitary adenomas (FIPA) and mutations in the aryl hydrocarbon receptor interacting protein (AIP) gene. Endocrinol. Metab. Clin. North Am. 2015. 44(1). 19-25. doi: 10.1016/j.ecl.2014.10.002.
- Chahal H.S., Chapple J.P., Frohman L.A., Grossman A.B., Korbonits M. Clinical, genetic and molecular characterization of patients with familial isolated pituitary adenomas (FIPA). Trends Endocrinol. Metab. 2010. 21(7). 419-27. doi: 10.1016/j.tem.2010.02.007.
- Nadhamuni V.S., Korbonits M. Novel Insights into Pituitary Tumorigenesis: Genetic and Epigenetic Mechanisms. Endocr. Rev. 2020. 41(6). 821-846. doi: 10.1210/endrev/bnaa006.
- Beckers A., Aaltonen L.A., Daly A.F., Karhu A. Familial isolated pituitary adenomas (FIPA) and the pituitary adenoma predisposition due to mutations in the aryl hydrocarbon receptor interacting protein (AIP) gene. Endocr. Rev. 2013. 34(2). 239-77. doi: 10.1210/er.2012-1013.
- Gadelha M.R., Trivellin G., Hernández Ramírez L.C., Korbonits M. Genetics of pituitary adenomas. Front. Horm. Res. 2013. 41. 111-40. doi: 10.1159/000345673.
- Feki M.M., Mnif F., Kamoun M., Charfi N., Rekik N., Naceur B.B., Mnif L. et al. Ectopic secretion of GHRH by a pancreatic neuroendocrine tumor associated with an empty sella. Ann. Endocrinol. (Paris). 2011. 72(6). 522-5. doi: 10.1016/j.ando.2011.06.002.
- Biswal S., Srinivasan B., Dutta P., Ranjan P., Vaiphei K., Singh R.S., Thingnam S.S. Acromegaly caused by ectopic growth hormone: a rare manifestation of a bronchial carcinoid. Ann. Thorac. Surg. 2008. 85(1). 330-2. doi: 10.1016/j.athoracsur.2007.06.072.
- Mankin H.J., Jupiter J., Trahan C.A. Hand and foot abnormalities associated with genetic diseases. Hand (N Y). 2011. 6(1). 18-26. doi: 10.1007/s11552-010-9302-8.
- Vandeva S., Tichomirowa M.A., Zacharieva S., Daly A.F., Beckers A. Genetic factors in the development of pituitary adenomas. Endocr. Dev. 2010. 17. 121-133. doi: 10.1159/000262534.
- Borson-Chazot F., Garby L., Raverot G., Claustrat F., Raverot V., Sassolas G. Acromegaly induced by ectopic secretion of GHRH: a review 30 years after GHRH discovery. Ann. Endocrinol. (Paris). 2012. 73(6). 497-502. doi: 10.1016/j.ando.2012.09.004.
- Boikos S.A., Stratakis C.A. Carney complex: Pathology and molecular genetics. Neuroendocrinology. 2006. 83(3–4). 189-99. doi: 10.1159/000095527.
- Raverot G., Arnous W., Calender A., Trouillas J., Sassolas G., Bournaud C., Pugeat M. et al. Familial pituitary adenomas with a heterogeneous functional pattern: clinical and genetic features. J. Endocrinol. Invest. 2007. 30(9). 787-90. doi: 10.1007/BF03350819.
- You C., Qiao F., Jiang S., Xiao A. Growth hormone secreting pituitary adenoma associated with Rathke's cleft cyst. Neurol. India. 2012. 60(3). 310-1. doi: 10.4103/0028-3886.98521.
- Phillips J.D., Yeldandi A., Blum M., Hoyos A. Bronchial carcinoid secreting insulin-like growth factor-1 with acromegalic features. Ann. Thorac. Surg. 2009. 88(4). 1350-2. doi: 10.1016/j.athoracsur.2009.02.042.
- Caimari F., Korbonits M. Novel Genetic Causes of Pituitary Adenomas. Clin. Cancer. Res. 2016. 22(20). 5030-5042. doi: 10.1158/1078-0432.CCR-16-0452.
- Pepe S., Korbonits M., Iacovazzo D. Germline and mosaic mutations causing pituitary tumours: genetic and molecular aspects. J. Endocrinol. 2019. 240(2). R21-R45. doi: 10.1530/JOE-18-0446.
- Tatsi C., Stratakis C.A. The Genetics of Pituitary Adenomas. J. Clin. Med. 2019. 9(1). 30. doi: 10.3390/jcm9010030.
- Ozcan-Kara P., Mahmoudian B., Erbas B., Erbas T. McCune-Albright syndrome associated with acromegaly and bipolar affective disorder. Eur. J. Intern. Med. 2007. 18(8). 600-2. doi: 10.1016/j.ejim.2007.02.030.
- Sung H.S., Yoon H.D., Shon H.S., Kim H.T., Choi W.Y., Seo C.J., Lee J.H. A case of McCune-Albright syndrome with associated multiple endocrinopathies. Korean J. Intern. Med. 2007. 22(1). 45-50. doi: 10.3904/kjim.2007.22.1.45.
- Collins M.T., Singer F.R., Eugster E. McCune-Albright syndrome and the extraskeletal manifestations of fibrous dysplasia. Orphanet. J. Rare Dis. 2012. 7(suppl. 1). S4. doi: 10.1186/1750-1172-7-S1-S4.
- Salenave S., Boyce A.M., Collins M.T., Chanson P. Acromegaly and McCune-Albright syndrome. J. Clin. Endocrinol. Metab. 2014. 99(6). 1955-69. doi: 10.1210/jc.2013-3826.
- Zatelli M.C., Tagliati F., Ruvo M.D., Castermans E., Cavazzini L., Daly A.F., Ambrosio M.R. et al. Deletion of exons 1–3 of the MEN1 gene in a large Italian family causes the loss of menin expression. Fam. Cancer. 2014. 13(2). 273-80. doi: 10.1007/s10689-014-9702-y.
- Kamilaris C.D.C., Stratakis C.A. Multiple Endocrine Neoplasia Type 1 (MEN1): An Update and the Significance of Early Genetic and Clinical Diagnosis. Front. Endocrinol. (Lausanne). 2019. 10. 339. doi: 10.3389/fendo.2019.00339.
- Lemos M.C., Thakker R.V. Multiple endocrine neoplaslia type 1 (MEN 1): Analysis of 1336 mutations reported in the first decade following identification of the gene. Hum. Mutat. 2008. 29(1). 22-32. doi: 10.1002/humu.20605.
- Thakker R.V., Newey P.J., Walls G.V., Bilezikian J., Dralle H., Ebeling P.R., Melmed S. et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J. Clin. Endocrinol. Metab. 2012. 97(9). 2990-3011. doi: 10.1210/jc.2012-1230.
- Espiard S., Bertherat J. Carney complex. Front. Horm. Res. 2013. 41. 50-62. doi: 10.1159/000345669.
- Cazabat L., Ragazzon B., Groussin L., Bertherat J. PRKAR1A mutations in primary pigmented nodular adrenocortical disease. Pituitary. 2006. 9(3). 211-9. doi: 10.1007/s11102-006-0266-1.
- Schamun M.B.B., Correa R., Graffigna P., Miguel V., Day P.F. Carney complex review: Genetic features. Endocrinol. Diabetes Nutr. 2018. 65(1). 52-59. doi: 10.1016/j.endinu. 2017.09.006.
- Matyakhina L., Pack S., Kirschner L.S., Pak E., Mannan P., Jaikumar J., Taymans S.E. et al. Chromosome 2 (2p16) abnormalities in carney complex tumours. J. Med. Genet. 2003. 40(4). 268-77. doi: 10.1136/jmg.40.4.268.
- Forlino A., Vetro A., Garavelli L., Ciccone R., London E., Stratakis C.A., Zuffardi O. PRKACB and Carney complex [Text]. N. Engl. J. Med. 2014. 13. 370(11). 1065-7. doi: 10.1056/NEJMc1309730.
- Stratakis C.A., Kirschner L.S., Carney J.A. Clinical and molecular features of the Carney complex: diagnostic criteria and recommendations for patient evaluation. J. Clin. Endocrinol. Metab. 2001. 86(9). 4041-6. doi: 10.1210/jcem.86.9.7903.
- Roy A.N.S., Radin M., Sarabi D., Shaoulian E. Familial recurrent atrial myxoma: Carney’s complex. Clin. Cardiol. 2011. 34(2). 83-6. doi: 10.1002/clc.20845.
- Kamilaris C.D.C., Faucz F.R., Voutetakis A., Stratakis C.A. Carney Complex. Exp. Clin. Endocrinol. Diabetes. 2019. 127(2–03). 156-164. doi: 10.1055/a-0753-4943.
- Malicka J., Świrska J., Nowakowski A. Familial acromegaly — case study of two sisters with acromegaly. Endokrynol. Pol. 2011. 62(6). 554-7.
- Beckers A., Daly A.F. The clinical, pathological, and genetic features of familial isolated pituitary adenomas. Eur. J. Endocrinol. 2007. 157(4). 371-82. doi: 10.1530/EJE-07-0348.
- Aaltonen L.A. Aryl hydrocarbon receptor-interacting protein and acromegaly. Horm. Res. 2007. 68(suppl. 5). 127-31. doi: 10.1159/000110607.
- Rostomyan L., Beckers A. Screening for genetic causes of growth hormone hypersecretion. Growth Horm. IGF Res. 2016. 30-31. 52-57. doi: 10.1016/j.ghir.2016.10.004.
- Melmed S., Bronstein M.D., Chanson P., Klibanski A., Casanueva F.F., Wass J.A.H., Strasburger C.J. et al. Consensus Statement on acromegaly therapeutic outcomes. Nat. Rev. Endocrinol. 2018. 14(9). 552-561. doi: 10.1038/s41574-018-0058-5.
- Leontiou C.A., Gueorguiev M., Spuy J., Quinton R., Lolli F., Hassan S., Chahal H.S. et al. The role of the aryl hydrocarbon receptor-interacting protein gene in familial and sporadic pituitary adenomas. J. Clin. Endocrinol. Metab. 2008. 93(6). 2390-401. doi: 10.1210/jc.2007-2611.
- Igreja S., Chahal H.S., King P., Bolger G.B., Srirangalingam U., Guasti L., Chapple J.P. et al. Characterization of aryl hydrocarbon receptor interacting protein (AIP) mutations in familial isolated pituitary adenoma familiess. Hum. Mutat. 2010. 31(8). 950-60. doi: 10.1002/humu.21292.
- Hernández-Ramírez L.C., Martucci F., Morgan R.M.L., Trivellin G., Tilley D., Ramos-Guajardo N., Iacovazzo D. et al. Rapid Proteasomal Degradation of Mutant Proteins Is the Primary Mechanism Leading to Tumorigenesis in Patients With Missense AIP Mutations. J. Clin. Endocrinol. Metab. 2016. 101(8). 3144-54. doi: 10.1210/jc.2016-1307.
- Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J., Grody W.W. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015. 17(5). 405-24. doi: 10.1038/gim.2015.30.
- Starker L.F., Akerström T., Long W.D., Delgado-Verdugo A., Donovan P., Udelsman R., Lifton R.P. et al. Frequent germ-line mutations of the MEN1, CASR, and HRPT2/CDC73 genes in young patients with clinically non-familial primary hyperparathyroidism. Horm. Cancer. 2012. 3(1–2). 44-51. doi: 10.1007/s12672-011-0100-8.
- Pardi E., Borsari S., Saponaro F., Bogazzi F., Urbani C., Mariotti S., Pigliaru F. et al. Mutational and large deletion study of genes implicated in hereditary forms of primary hyperparathyroidism and correlation with clinical features. PLoS One. 2017. 16. 12(10). e0186485. doi: 10.1371/journal.pone.0186485.
- Kihara M., Miyauchi A., Ito Y., Yoshida H., Miya A., Kobayashi K., Takamura Y. et al. MEN1 gene analysis in patients with primary hyperparathyroidism: 10-year experience of a single institution for thyroid and parathyroid care in Japan. Endocr. J. 2009. 56(5). 649-56. doi: 10.1507/endocrj.k08e-265.
- Sato M., Kihara M., Nishitani A., Murao K., Kobayashi S., Miyauchi A., Takahara J. Large and asymptomatic pancreatic islet cell tumor in a patient with multiple endocrine neoplasia type 1-Endocrine. 2000. 13(3). 263-6. doi: 10.1385/ENDO:13:3:263.
- Miyauchi A., Sato M., Matsubara S., Ohye H., Kihara M., Matsusaka K., Nishitani A. et al. A family of MEN1 with a novel germline missense mutation and benign polymorphisms. Endocr. J. 1998. 45(6). 753-9. doi: 10.1507/endocrj.45.753.
- Vierimaa O., Georgitsi M., Lehtonen R., Vahteristo P., Kokko A., Raitila A., Tuppurainen K. et al. Pituitary adenoma predisposition caused by germline mutations in the gene. Science. 2006. 312(5777). 1228-30. doi: 10.1126/science.1126100.
- Iwata T., Yamada S., Mizusawa N., Golam H.M., Sano T., Yoshimoto K. The aryl hydrocarbon receptor-interacting protein gene is rarely mutated in sporadic GH-secreting adenomas. Clin. Endocrinol. (Oxf). 2007. 66(4). 499-502. doi: 10.1111/j.1365-2265.2007.02758.x.
- Korbonits M., Storr H., Kumar A.V. Familial pituitary adenomas — who should be tested for AIP mutations? Clin. Endocrinol. (Oxf). 2012. 77(3). 351-6. doi: 10.1111/j.1365-2265.2012.04445.x.
- Personnier C., Cazabat L., Bertherat J., Gaillard S., Souberbielle J.C., Habrand J.L., Dufour C. et al. Clinical features and treatment of pediatric somatotropinoma: case study of an aggressive tumor due to a new AIP mutation and extensive literature review. Horm. Res. Paediatr. 2011. 75(6). 392-402. doi: 10.1159/000327831.
- Joshi K., Daly A.F., Beckers A., Zacharin M. Resistant Paediatric Somatotropinomas due to AIP Mutations: Role of Pegvisomant. Horm. Res. Paediatr. 2018. 90(3). 196-202. doi: 10.1159/000488856.