
Международный неврологический журнал 1(1) 2005
Вернуться к номеру
Миастения детского возраста: дифференциально-диагностические критерии аутоиммунной и врожденной форм
Авторы: Е.Ю. Мененкова, отдел нервно-мышечной патологии человека НИИ общей патологии и патофизиологии РАМН, Центральная клиническая больница МПС РФ, Москва
Рубрики: Неврология
Разделы: Клинические исследования
Версия для печати
Миастения с дебютом в детском возрасте составляет от 10% до 24% всех больных миастенией (Лобзин В.С., 1960; Гехт Б.М., 1974; Fenichel G.M., 1978; Snead O.C. et al., 1980; Szobоr A. et al., 1989; Herrmann D.N. et al., 1998; Morita Md. et al., 2001). Миастения относится к аутоиммунным заболеваниям, проявляется слабостью и патологической утомляемостью различных мышечных групп вследствие поражения нервно-мышечного синапса антителами к ацетилхолиновым рецепторам (АХР). Эти антитела являются иммуноглобулинами, обладающими высоким сродством к различным субъединицам рецептора. Вместе с тем, существует группа врожденных миастенических синдромов, клинические проявления которых в значительной степени близки к выявляемым у больных аутоиммунной миастенией, однако имеющим другой патогенетический механизм. В настоящее время известно, что в основе патогенеза врожденных миастенических синдромов лежат мутации генов, определяющие изменения структуры и функции субъединиц АХР (Engel A.G. et al., 1993; 1996; 1999; Ohno K. et al., 1999; 2000).
Важно подчеркнуть и то обстоятельство, что диагностика миастении в детском возрасте должна быть проведена до назначения патогенетической терапии, так как традиционные методы лечения аутоиммунной формы заболевания (глюкокортикоидные, иммуносупрессорные препараты, иммуноглобулины, плазмаферез и тимэктомия) не применяются при врожденной форме.
В связи с этим, целью данного исследования явилось изучение клинического паттерна детской аутоиммунной и врожденной миастении, а также особенностей нарушения нервно-мышечной передачи и организации потенциалов двигательных единиц при этих формах заболевания.
Материалы и методы
Обследовано 36 детей, больных аутоиммунной миастенией (1-я группа). Все больные были в возрасте от 1-го года до 13-ти лет, из них девочек — 21 (58,3%), мальчиков — 15 (41,7%) Соотношение детей женского и мужского пола составило 1,4:1. Средний возраст детей к началу заболевания составил 8,88 лет ± 3,76. Средний возраст девочек составил 10,19 ± 3,41 лет, мальчиков — 7,07 ± 3,53 лет. Также обследовано 24 больных с конгенитальными формами миастении (2-я группа). Возраст больных составил от 2-х месяцев до 12-ти лет. Средний возраст пациентов — 2,62 ± 3,05 лет. Среди больных было 13 девочек (54,17%), мальчиков — 11 (45,83%). Соотношение больных женского и мужского пола составило 1,2:1.
Контрольную группу (3-я группа) составили 38 больных аутоиммунной миастений в возрасте от 15-ти до 59-ти лет, из них — 32 (84,2%) женщины в возрасте от 15-ти до 59-ти лет, 6 (15,8%) мужчин в возрасте от 41-го до 59-ти лет. Соотношение больных женского и мужского пола составило 5,3:1. Средний возраст пациентов был 34,43±13,18 лет. Средний возраст женщин составил 31,4±11,29 лет, мужчин — 54,4±3,91 лет.
Клинический анализ включал оценку неврологического статуса и выраженности симптомов болезни: легкая степень тяжести, средняя и тяжелая степень.
Для оценки степени вовлечения в патологический процесс отдельных мышц туловища и конечностей была использована 4-балльная шкала, где 0 соответствует отсутствию вовлечения в патологический процесс данной мышцы (соответствует силе 5 баллов), 1 — минимальному вовлечению (соответствует силе 4 балла), 2 — умеренному вовлечению (соответствует силе 3 балла), 3 — тяжелому поражению мышцы (соответствует силе 2 балла и менее). Кроме того, больные были обследованы на наличие мышечных атрофий, гипотонии, снижения сухожильных рефлексов.
Электромиографическое исследование было проведено в дельтовидной мышце.
Все обследования проводились на фоне отмены антихолинэстеразных препаратов не менее чем за 6-8 часов до начала исследования. Электомиографические исследования проводились на компьютерных электромиографах «NEUROPAK-2» производства Японии и «Нейромиан-МЕДИКОМ» производства России.
Исследование функционального состояния нервно-мышечной передачи включало: 1) изучение величины амплитуды (площади) негативной фазы М-ответа в ответ на одиночный супрамаксимальный стимул; 2) исследование величины декремента амплитуды (площади) М-ответа при стимуляции мышцы частотой 3 имп/с в процентах по отношению 5-го М-ответа к 1-му по формуле:
Декремент (%) = [100 – (потенциал 5/ потенциал 1) × 100] %.
Исследование изменения амплитуды (площади) М-ответа при стимуляции частотой 3 имп/с через 10 с после окончания максимального произвольного усилия (в течение 10 с) (постактивационное облегчение — ПАО) — в процентах по отношению к исходному М-ответу и 5-го М-ответа к 1-му.
Исследование потенциалов двигательных единиц (ПДЕ) проводилось не всем пациентам, и чаще всего, если имелись подозрения о сочетании миастении с другими заболеваниями: 24 больных контрольной группы, 24 больных детской миастенией, а так же 20 больных врожденной миастенией для исключения мышечного и невритического компонентов заболевания. Потенциалы двигательных единиц изучались в дельтовидной мышце. Исследовалась средняя длительность ПДЕ. Также оценивалась средняя амплитуда ПДЕ (мкв), форма ПДЕ и спонтанная активность мышечных волокон и двигательных единиц.
Результаты и обсуждение
Данные, полученные в настоящем исследовании, позволяют обсудить целый ряд вопросов, связанных с патогенезом миастенических нарушений в детском возрасте, и, в первую очередь, особенности двигательных расстройств у больных с детской аутоиммунной и врожденной формами миастении.
У обследованных нами больных детской аутоиммунной миастенией был выявлен ряд клинических особенностей, который включал более частое поражение экстраокулярной мускулатуры (72,2% детей, p<0,01), мышц шеи (77,8% детей, p<0,01), дельтовидной (100,0% детей, p<0,001) и подвздошно-поясничной мыщц (94,4 % детей, р<0,01), а также дистальных мышц кисти (75,0% детей, р<0,001) и стопы (41,7% детей, p<0,05), более частое поражение дыхательной мускулатуры (30,6% детей, p<0,05), генерализованное снижение мышечного тонуса по сравнению со взрослыми пациентами (52,8% детей, p<0,001).
Также было выявлено, что миастения у детей отличается от миастении взрослых степенью выраженности симптомов. У больных детской миастенией был достоверно более выраженный птоз (р<0,05), а также более значительное снижение силы круговой мышцы глаза (р<0,05), мышц шеи (р<0,005), дельтовидной (р<0,001), трехглавой мышцы плеча (р<0,05), подвздошно-поясничной мышцы (р<0,005), мышц кисти р<0,001) и стопы (р<0,05) и более выраженные нарушения дыхания (р<0,01) (см. таблицу).
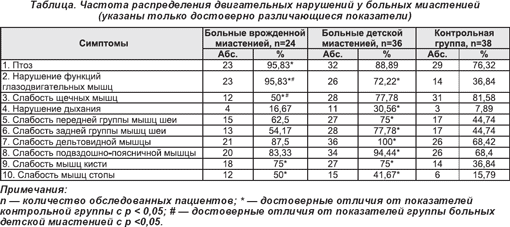
В нашем исследовании у больных врожденной миастенией выявлены следующие клинические особенности: частое наличие птоза (98,5%; р<0,05) и офтальмопареза (95,8%; р<0,001), относительно менее частое поражение мимической мускулатуры (50,0%; р<0,01), более легкий бульбарный синдром, частое поражение дистальных мышц кистей (75,0%; р<0,01) и стоп (50%; р<0,001) с наличием амиотрофий (50,0%; р<0,001), мышечной гипотонии (75,0%; р<0,001) и сниженных сухожильных рефлексов (37,5%; р<0,05).
Больные врожденной миастенией статистически достоверно отличались от больных аутоиммунной миастенией (контрольная группа) более выраженным птозом (р<0,005), тяжелым поражением глазодвигательных мышц (р<0,001), дельтовидной мышцы (р<0,05), мышц кисти (р<0,001) и стопы (р< 0,005). От больных детской миастенией больные врожденной формой заболевания отличались тяжелым поражением глазодвигательных мышц (р<0,001) и легкими нарушениями фонации (р<0,05).
Различия клинических проявлений миастении детского возраста, определяемые паттернами аутоиммунной и врожденной форм заболевания, обусловлены повреждением различных звеньев механизма, осуществляющего передачу возбуждения с нерва на мышцу, а в случаях патологии одного и того же звена — отличием этиологической причины, приводящей к этому повреждению. Так, патология холинорецептора при детской аутоиммунной миастении связана с наличием специфических аутоантител, тогда как при врожденных формах — генетически обусловлена нарушением числа рецепторов или аномалией их функционирования.
Наиболее трудно провести дифференциальный диагноз между детской аутоиммунной и врожденной миастенией у тех пациентов, у которых симптомы заболевания не были оценены в период новорожденности или в раннем детском периоде. Так как даже на современном уровне знаний генетическое обследование пациентов остается малодоступным методом исследования в нашей стране, а титры антител к АХР пациентов обеих групп чаще оказываются отрицательными, то электрофизиологические методы могут помочь в установлении правильного диагноза.
На основании проведенного нами исследования было выявлено, что при более выраженной степени снижения силы дельтовидной мышцы у больных детской аутоиммунной миастенией электрофизиологические характеристики, отражающие состояние нервно-мышечной передачи, у этой группы пациентов не отличаются от таковых у больных аутоиммунной миастенией контрольной группы. Так, среднее значение декремента амплитуды М-ответа в дельтовидной мышце у больных детской миастенией составило –39,14±22,23%, а в контрольной группе — –33,74±20,96%.
Проведенное сопоставление электромиографических показателей у больных детской аутоиммунной и врожденной миастенией показало, что при равном значительном снижении силы мышцы величина декремента амплитуды М-ответа была достоверно больше (р<0,001) у больных детской миастенией (–39,14±22,23%), чем у больных врожденной миастенией (–16,04± 14,67%). Других различий электромиографических показателей между больными детской и врожденной миастенией не было выявлено.
Приведенные в нашей работе данные клинических и электромиографических характеристик, отражающих состояние нервно-мышечной передачи у больных в трех сравниваемых группах, показали, что больные врожденной миастенией отличаются от больных детской миастенией и взрослых больных аутоиммунной миастенией значительно меньшей величиной декремента амплитуды М-ответа при равной степени снижения силы в проксимальной (дельтовидной) мышце.
Известно, что нарушение структуры двигательной единицы ведет к увеличению числа полифазных потенциалов двигательных единиц (ПДЕ). У больных с аутоиммунной миастенией в 76% мышц выявляется более одного полифазного ПДЕ, а в некоторых мышцах их число достигает 50-70%, средняя величина составляет 24,2±19,3% (Гехт Б.М. и др., 1997).
В нашем исследовании у больных всех трех групп количество полифазных потенциалов в дельтовидной мышце превышало 5% и составляло у больных детской миастенией 36,04±25,41%, у больных врожденной миастенией — 43,5±21,64% и у больных контрольной группы — 27,71±16,35%. Приведенные результаты свидетельстуют о том, что среднее значение количества полифазных ПДЕ в дельтовидной мышце у больных врожденной миастенией достоверно выше (р<0,05) этого же показателя у больных контрольной группы. Увеличение количества полифазных двигательных единиц и потенциалов с увеличенной амплитудой и длительностью в мышцах больных с врожденными формами миастении свидетельствует о компенсаторной перестройке нейромоторного аппарата в условиях хронического дефицита влияния синаптического передатчика на мышечное волокно.
Выявление клинических и электрофизиологических критериев дифференциации различных клинических форм миастении с дебютом в детском возрасте позволяет практическому врачу на основании данных неврологического статуса и рутинных методов электромиографического обследования предположить наличие аутоиммунной или врожденной формы заболевания и назначить адекватное лечение, а также направить пациентов в миастенический центр для проведения дополнительных исследований, подтверждающих данное предположение.
1. Гехт Б.М. Синдромы патологической мышечной утомляемости. — М., Медицина, 1974. — 200 c.
2. Гехт Б.М., Касаткина Л.Ф., Самойлов М.И., Санадзе А.Г. Электромиография в диагностике нервно-мышечных заболеваний. — Таганрог: Издательство ТРТУ, 1997. — 370 с.
3. Лобзин В.С. Миастения. — Л., Медгиз, 1960. — 155 с.
4. Engel A.G., Nagel A., Walls T.J. Congenital Myasthenic Syndromes: 1.Deficiency and short open time of the acetylcholine receptor // Muscle Nerve. — 1993. — V.16. — P. 1284-1292.
5. Engel A.G., Ohno K., Milone M. et al. New mutations in acetylcholine receptor subunit genes reveal heterogeneity in the slow-channel congenital myasthenic syndrome // Hum. Mol. Genet. — 1996. — V. 5.—1217-1227.
6. Engel A.G., Ohno K., Sine S.M. Congenital myasthenic syndromes: recent advances // Arch. Neurol. — 1999. — V. 56(2). — P.163-167.
7. Fenichel G.M. Clinical syndrome of myasthenia in infancy and childhood // Arch. Neurol. — 1978. — V. 35. — P. 97-103.
8. Herrmann D.N., Carney P.R., Wald J.J. Juvenile myasthenia gravis: treatment with immuneglobuline and thymectomy // Pediatr. Neurol. — 1998. — V. 18(1). — P. 63-66.
9. Ludin H.P. Electromyography in practice // Stuttgart. — 1980. — 174 p.
10. Morita Md., Gabbai A.A., Oliveira A.S., Penn A.S. Myasthenia gravis in children: analysis of 18 patients // Arq. Neuropsiquiatr. — 2001. — V. 59(3-B). — P. 681-685.
11. Ohno K., Brengman J.M., Felice K.J., Comblath D.R., Engel A.G. Congenital endplate acetylcholinesterase deficiency caused by a nonsense mutation and an A-to-G splice site mutation at position +3 of the collagen-like tail subunit gene (COLQ): How does G at position +3 result in aberrant splicing? // Am. J. Hum. Genet. — 1999. — V. 65. — P. 635-644.
12. Ohno K., Engel A.G., Brengman J.M., et al. The spectrum of mutations causing endplate acetylcholinesterase deficiency // Ann. Neurol. — 2000. — V. 47. — P. 162-170.
13. Snead O.C., Benton J.W., Dwyer D., et al. Juvenile myasthenia gravis // Neurology. — 1980. — V. 30. — P. 732-739.
14. Szobor A., Mattyus A., Molnar J. Myasthenia gravis in childhood and adolescence: report on 209 patients and review of literature // Acta. Pediatr. Hung. — 1988-1989. — V. 29. — P. 299-312.