
Международный неврологический журнал 4 (66) 2014
Вернуться к номеру
Болезнь Паркинсона и паркинсонические синдромы (лекция)
Авторы: Евтушенко С.К., Головченко Ю.И., Труфанов Е.А. - Донецкий национальный медицинский университет имени М. Горького; Национальная медицинская академия последипломного образования имени П.Л. Шупика, г. Киев; Луганский государственный медицинский университет
Рубрики: Неврология
Разделы: Клинические исследования
Версия для печати
Лекция посвящена одному из труднокурабельных заболеваний нервной системы — болезни Паркинсона и ее фенокопиям. В лекции рассматривается актуальность проблемы, которая исходит из высокой инвалидизации больных. На основе мировой литературы и многих исследований авторы излагают особенности клиники болезни Паркинсона и приводят ее тщательную дифференциальную диагностику.
Лекція присвячена одному з тяжкокурабельних захворювань нервової системи — хворобі Паркінсона і її фенокопіям. У лекції розглядається актуальність проблеми, що виходить із високої інвалідизації хворих. На основі світової літератури та багатьох досліджень автори викладають особливості клініки хвороби Паркінсона й наводять її ретельну диференціальну діагностику.
The lecture deals with one of hard to treat diseases of the nervous system — Parkinson’s disease and its phenocopies. The lecture discusses the relevance of the problem, which is based on the high disability of patients. On the basis of world literature and many studies the authors describe clinical features of Parkinson’s disease and provide its careful differential diagnosis.
болезнь Паркинсона, прогрессирующий надъядерный паралич, системная атрофия, деменция с тельцами Леви, кортикобазальная дегенерация, сосудистый паркинсонизм.
хвороба Паркінсона, прогресуючий над’ядерний параліч, системна атрофія, деменція з тільцями Леві, кортикобазальна дегенерація, судинний паркінсонізм.
Parkinson’s disease, progressive supranuclear palsy, system atrophy, dementia with Lewy bodies, corticobasal degeneration, vascular parkinsonism.
Статья опубликована на с. 16-31
Болезнь Паркинсона (БП) — прогрессирующее инвалидизирующее неврологическое заболевание, возникающее в результате дегенеративных изменений в допаминергических нейронах. Это довольно распространенное заболевание развивается в среднем у 100–329 человек на 100 тысяч населения и клинически характеризуется четырьмя главными признаками: тремором покоя, брадикинезией, ригидностью и постуральной неустойчивостью, а также наличием широкого спектра других двигательных и недвигательных проявлений. Несмотря на то, что болезнь Паркинсона является распространенным заболеванием, установление клинического диагноза нередко является затруднительным, особенно на ее ранних стадиях.
Количество зарегистрированных случаев болезни Паркинсона в странах Европейского Союза составляет от 112 случаев на 100 000 населения в Швеции до 229,3 случая в Италии. В Азии болезнь Паркинсона встречается с частотой от 118 (Япония) до 374 случаев (Южная Корея) на 100 000 населения, в США и Канаде зарегистрировано от 125 до 329 страдающих БП на 100 000 населения. В странах Африки зарегистрировано от 7 (Эфиопия) до 43 (Тунис) страдающих БП на 100 000 населения.
Низкое число зарегистрированных случаев болезни Паркинсона на территории Украины (61,4 случая на 100 000 населения) по сравнению с эпидемиологическими данными о болезни Паркинсона в странах Европейского Союза, Северной Америки, Азии и Австралии может быть связано с гиподиагностикой этого заболевания. Существенные различия в количестве зарегистрированных случаев болезни Паркинсона в различных регионах Украины (от 30,5 до 121,6 случая на 100 000 населения) подтверждают это предположение. К сожалению, в Украине отсутствуют статистические данные о распространенности других экстрапирамидных заболеваний (Труфанов Е.А., 2013).
От болезни Паркинсона следует отличать синдромы паркинсонизма (паркинсонизм-плюс): мультисистемную атрофию, прогрессирующий надъядерный паралич, деменцию с тельцами Леви, кортикобазальную дегенерацию, сосудистый паркинсонизм, марганцевый паркинсонизм и другие интоксикационные формы, которые отличаются наличием дополнительных признаков, более быстрым прогрессированием и низкой эффективностью или отсутствием эффекта от противопаркинсонических препаратов (табл. 1).
/17/17.jpg)
Диагностику болезни Паркинсона и паркинсонических синдромов существенно улучшает использование специальных критериев постановки диагноза при этих заболеваниях.
И все же золотым стандартом постановки диагноза при жизни больного является клинический метод. Клиническое обследование и длительное наблюдение являются наилучшим методом для подтверждения диагноза в течение всей жизни больного. Дальнейшие исследования необходимы для улучшения диагностической точности и прогнозирования прогрессирования заболевания.
Цель данной лекции — представить современные данные о диагностике болезни Паркинсона и его фенокопий и осветить наиболее значимые способы терапии данных заболеваний.
Материал и методы
При написании лекции использовались ресурсы PubMed (1990–2013 гг.) и UpToDate (2012 г.), а также материалы собственных многолетних наблюдений.
Результаты и их обсуждение
Болезнь Паркинсона
Болезнь Паркинсона — первое описанное и наиболее изученное заболевание экстрапирамидной системы. Несмотря на достигнутые успехи в медикаментозном и хирургическом лечении, болезнь Паркинсона остается неуклонно прогрессирующим и, в конечном счете, значительно инвалидизирующим заболеванием.
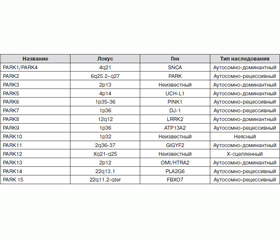
В 1997 году была обнаружена зависимость между наследственными аутосомно-доминантными формами болезни Паркинсона и синуклеиновой генетической мутацией. С тех пор была установлена взаимосвязь двух десятков различных генов (табл. 2) с наследственными формами болезни Паркинсона и повышенным риском развития этого заболевания.
/17/17_2.jpg)
Наследственный анамнез прослеживается приблизительно в 15–20 % случаев болезни Паркинсона, и в настоящее время считается, что подавляющее большинство случаев болезни Паркинсона являются идиопатическими. При наследственных формах болезни Паркинсона, как правило, отмечают более ранний возраст начала заболевания.
Клинические проявления. Средний возраст начала болезни Паркинсона составляет около 60 лет, при этом до 5 % случаев болезни Паркинсона начинаются в возрасте до 40 лет.
Современные исследования показали, что преклинический период болезни Паркинсона в среднем составляет от пяти до двадцати лет. Считается, что преклинический период болезни Паркинсона начинается с поражения обонятельных луковиц, передних обонятельных ядер и дорсального ядра блуждающего нерва.
К предвестникам болезни Паркинсона относятся беспричинное ухудшение обоняния, яркие и живые сновидения, депрессия и запоры. Вышеперечисленные симптомы могут возникать за много лет до двигательных проявлений болезни Паркинсона. Эти недвигательные проявления болезни Паркинсона в будущем могут помочь диагностировать заболевание на его премоторной стадии, что окажет помощь в раннем использовании разработанных лекарственных препаратов, способных модифицировать течение заболевания.
Первоначальные моторные симптомы болезни Паркинсона обычно начинаются на одной стороне тела и появляются на другой его стороне только через 2–5 лет заболевания. Асимметричное начало двигательных симптомов является одним из самых надежных диагностических факторов как спорадических, так и наследственных форм болезни Паркинсона.
И все же основным клиническим проявлением болезни Паркинсона является брадикинезия, представляющая собой замедленность движений вследствие затруднения их планирования, инициирования и выполнения. На начальных стадиях заболевания брадикинезия более заметна при выполнении мелких движений, что в первую очередь вызывает затруднения при одевании, еде, выполнении гигиенических процедур и письме. Отмечается снижение амплитуды маятникообразного движения рук при ходьбе. По мере прогрессирования заболевания присоединяются и нарастают другие проявления брадикинезии (медленная ходьба, гипомимия, брадилалия и др.).
К другим классическим проявлениям болезни Паркинсона относятся тремор, ригидность, постуральная неустойчивость, шаркающая походка, сгибательная поза и застывания при ходьбе.
Тремор. Тремор покоя наблюдается у 80 % страдающих болезнью Паркинсона, и он бывает ее первоначальным двигательным симптомом у 40 % больных.
Для болезни Паркинсона обычно характерен асимметричный тремор кистей одной или обеих рук в покое по типу «счета монет» или «скатывания пилюль» частотой 4–6 Гц, исчезающий при выполнении движений руками. Также может наблюдаться тремор ног в покое, тремор подбородка и редко — тремор головы. При болезни Паркинсона иногда наблюдается и постуральный тремор рук, который может начинаться за много лет до других проявлений заболевания.
В тех случаях, когда у больного вместе с постуральным тремором наблюдаются минимальные (или сомнительные) проявления брадикинезии, диагноз болезни Паркинсона может вызывать серьезные затруднения.
Моторные симптомы болезни Паркинсона в настоящее время рассматриваются в качестве важных элементов в клиническом спектре заболевания. Моторные симптомы болезни Паркинсона (так же, как и двигательные симптомы) достаточно часто вызывают выраженную инвалидизацию и существенно снижают качество жизни больных. К немоторным симптомам болезни Паркинсона относятся разнообразные психические, вегетативные, чувствительные и другие расстройства. При посещении невролога недвигательные симптомы не всегда распознаются и точно диагностируются.
Все страдающие болезнью Паркинсона должны быть опрошены на предмет наличия моторных симптомов, так как доступно симптоматическое лечение многих из этих симптомов. Вместе с тем некоторые из моторных проявлений могут быть частью off-периодов, в связи с чем полезна коррекция допаминергической терапии.
Раннее распознавание моторных проявлений болезни Паркинсона и вызванной ими инвалидизации не только имеет важное значение для улучшения функционального состояния больных, но и способствует расширению знаний о природе нейродегенеративного процесса при болезни Паркинсона.
В связи с этим чрезвычайно важно использование карты самочувствия при болезни Паркинсона — Parkinson’s Well-Being Map (WBM), предназначенной для самооценки симптомов заболевания. Приоритет внедрения данной методики в Украине принадлежит проф. И.Н. Карабань (2011–2013 гг.).
К психическим расстройствам при болезни Паркинсона относятся когнитивные нарушения, брадифрения, депрессия, апатия, тревога, потеря интересов и инициативы, повышенная утомляемость, нарушения сна, зрительные галлюцинации, бред и психозы. К нарушениям сна при болезни Паркинсона — поведенческие расстройства фазы быстрого сна, яркие и живые сновидения, чрезмерная дневная сонливость, апноэ во сне, синдром беспокойных ног.
В отличие от болезни Альцгеймера, при которой наблюдается корковая деменция, у страдающих болезнью Паркинсона обычно развивается подкорковый тип деменции. Ряд психических расстройств может развиваться как побочный эффект допаминергических препаратов (в первую очередь агонистов допамина). К побочным эффектам допаминергических препаратов можно отнести различные расстройства импульсного контроля: гемблинг, компульсивный шоппинг, гиперсексуальность.
Степень когнитивных нарушений у страдающих болезнью Паркинсона коррелирует с возрастом начала заболевания. Имеется достоверная связь между показателями когнитивных функций и тяжестью нарушений речи, а также постуральной устойчивостью.
Частыми инвалидизирующими осложнениями болезни Паркинсона являются вегетативные нарушения, которые существенно влияют на качество жизни больного и его повседневную активность, а в некоторых случаях могут приводить к смерти. К вегетативным нарушениям при болезни Паркинсона относятся ортостатическая гипотензия, расстройства мочеиспускания (учащенное, задержки, недержание), запоры, эректильная дисфункция, чрезмерное потоотделение, себорейный дерматит и др.
Существует мнение, что возможна зависимость между двигательными расстройствами, когнитивной дисфункцией и вегетативными нарушениями у страдающих болезнью Паркинсона. Следовательно, вегетативные нарушения и когнитивные расстройства могут служить потенциальными прогностическими факторами прогрессирования двигательных расстройств у таких больных.
Кроме того, лекарственные препараты, применяющиеся для лечения двигательных расстройств при болезни Паркинсона, иногда также могут вызывать или усиливать вегетативные нарушения, делая лечение болезни Паркинсона затруднительным.
Нарушения глотания являются частым проявлением болезни Паркинсона и паркинсонических синдромов особенно и обычно не поддаются воздействию противопаркинсонических препаратов. Аспирация пищи вследствие нарушения глотания может приводить к аспирационной пневмонии, которая является одним из наиболее частых сопутствующих заболеваний, приводящих к смерти, при болезни Паркинсона и паркинсонических синдромах.
Дисфагия обычно ассоциируется с тяжестью болезни Паркинсона.
Имеется взаимосвязь между нарушением глотания и качеством жизни у страдающих болезнью Паркинсона. В связи с этим идентификация нарушения глотания крайне необходима. Применение обычного рентгенологического теста не уточняет этиологию нарушения глотания.
Чувствительные расстройства. К чувствительным расстройствам при болезни Паркинсона относятся расстройства обоняния, боль и парестезии. Нарушения обоняния наблюдаются у 70–90 % страдающих болезнью Паркинсона, и обычно они развиваются за много лет до развития двигательных проявлений заболевания. Учитывая широкую распространенность нарушений обоняния уже на ранних стадиях заболевания, данный моторный симптом может использоваться при проведении дифференциальной диагностики болезни Паркинсона с паркинсоническими синдромами.
Дискинезия. Дискинезией называют непроизвольные движения, которые чаще всего возникают на фоне длительного лечения леводопой. Наиболее распространенным видом леводопа-индуцированной дискинезии является леводопа-индуцированная хорея.
Дискинезия является одним из самых существенных двигательных осложнений при использовании противопаркинсонических препаратов (леводопы), которое в ряде случаев значительно ухудшает качество жизни больных. Снижение дозировок леводопы приводит к уменьшению дискинезии и в то же время к нарастанию двигательных симптомов паркинсонизма и увеличению off-периодов, что еще больше ухудшает состояние страдающих болезнью Паркинсона. Индуцированная леводопой дискинезия развивается у трети страдающих болезнью Паркинсона в течение первых 2 лет лечения леводопой и больше чем у половины больных после 5 лет лечения.
Асимметрия двигательных синдромов также характерна для страдающих болезнью Паркинсона. В большинстве случаев наблюдается асимметрия двигательных симптомов, которая возникает при спорадических и наследственных формах болезни Паркинсона.
Клинические шкалы тяжести болезни Паркинсона. Для оценки тяжести болезни Паркинсона используются стандартные международные шкалы: UPDRS, MDS-UPDRS, шкала повседневной жизненной активности Schwab и England, модифицированная шкала Hoehn и Yahr.
Инструментальные методы диагностики. При проведении МРТ у страдающих болезнью Паркинсона обычно не обнаруживается патологических изменений или выявляются неспецифические изменения, но, несмотря на низкую чувствительность и специфичность МРТ головного мозга, проведение МРТ обязательно, поскольку это исследование может быть полезно в качестве дополнительного метода дифференциальной диагностики болезни Паркинсона и паркинсонических синдромов.
У 70,71 % обследованных лиц с болезнью Паркинсона с помощью МРТ не обнаруживается никаких патологических изменений в головном мозге. 12,8 % страдающих болезнью Паркинсона имеют атрофические изменения головного мозга, которые соответствуют их возрасту (2,8 %) или несколько более выражены, чем у здоровых людей этой возрастной группы (10,1 %), но у 15,0 % больных наблюдаются минимальные или нерезкие ишемические изменения («немые» инфаркты) в белом веществе головного мозга.
В то же время атрофические изменения головного мозга при проведении магнитно-резонансной томографии обнаруживаются у 64,7 % больных с мультисистемной атрофией, 20 % больных деменцией с тельцами Леви, 50 % больных с прогрессирующим надъядерным параличом, практически у всех больных с кортикобазальной дегенерацией и сосудистым паркинсонизмом. У больных с мультисистемной атрофией и прогрессирующим надъядерным параличом атрофические изменения более выражены в мозжечке, а у больных с кортикобазальной дегенерацией — в лобных долях головного мозга.
При визуализации транспортеров допамина (DaTSCAN) у страдающих болезнью Паркинсона обнаруживается значительное уменьшение транспортеров допамина в стриатуме, что непосредственно связано с уменьшением в нем допаминовых нигростриальных нервных окончаний. DaTSCAN позволяет с высокой точностью дифференцировать болезнь Паркинсона от эссенциального тремора, психогенного паркинсонизма и лекарственно-индуцированного паркинсонизма.
Такие методы диагностики, как позитронно-эмиссионная томография (ПЭТ) и однофотонная эмиссионная компьютерная томография (SPECT), особенно важны в дифференциальной диагностике болезни Паркинсона.
По результатам транскраниальной ультразвуковой допплерографии сосудов головного мозга изменения кровотока в вертебробазилярном бассейне выявляются у 58,3 % лиц с болезнью Паркинсона. Обнаруженные патологические изменения не специфичны для болезни Паркинсона, так как подобные изменения могут встречаться у людей той же возрастной группы без наличия какой-либо экстрапирамидной патологии.
Общепринятыми диагностическими критериями болезни Паркинсона в настоящее время являются клинические диагностические критерии Общества болезни Паркинсона Соединенного Королевства. Эти критерии состоят из 3 этапов (шагов).
Шаг 1. Устанавливается диагноз паркинсонизма, при котором обязательно должна присутствовать брадикинезия плюс один из перечисленных ниже синдромов: мышечная ригидность, тремор покоя, постуральная неустойчивость, не связанная с первичной зрительной, вестибулярной, мозжечковой или проприоцептивной дисфункцией. После того как диагноз паркинсонизма заподозрен и появилась необходимость провести дифференциальную диагностику между болезнью Паркинсона и паркинсоническими синдромами, переходят ко второму шагу диагностических критериев.
Шаг 2. Критерии исключения болезни Паркинсона:
— повторные инсульты в анамнезе со ступенеобразным прогрессированием паркинсонических признаков;
— повторные травмы головы в анамнезе;
— перенесенный энцефалит в анамнезе;
— окулогирные кризы;
— начало паркинсонических симптомов на фоне лечения нейролептиками;
— наличие более одного родственника с похожим заболеванием;
— длительная ремиссия;
— наличие только односторонних признаков после 3 лет болезни;
— надъядерный паралич взора;
— мозжечковые знаки;
— ранние выраженные вегетативные расстройства;
— ранняя выраженная деменция с расстройствами памяти, речи и праксиса;
— симптом Бабинского;
— наличие опухоли мозга или открытая гидроцефалия, выявленная при проведении нейровизуализационного обследования;
— отсутствие эффекта при приеме больших доз леводопы (при условии отсутствия мальабсорбции);
— больной подвергался воздействию MPTP.
Шаг 3. Позитивные проспективные критерии болезни Паркинсона (для постановки достоверного диагноза болезни Паркинсона необходимо три или более следующих критерия в сочетании с шагом 1):
— одностороннее начало болезни;
— наличие тремора покоя;
— прогрессирование болезни;
— сохраняющаяся асимметрия, более выраженные симптомы на стороне начала болезни;
— отличный терапевтический эффект леводопы (70–100 %);
— выраженная индуцированная леводопой хорея;
— терапевтический эффект леводопы в течение 5 и более лет;
— клиническое течение болезни в течение 10 и более лет.
Тест с одноразовым применением леводопы или апоморфина не следует использовать при проведении дифференциальной диагностики болезни Паркинсона и паркинсонических синдромов.
Точная диагностика болезни Паркинсона может быть довольно сложной задачей, особенно на ее ранних стадиях, так как основные проявления нейродегенеративных заболеваний могут совпадать. Особенно сложна дифференциальная диагностика болезни Паркинсона с паркинсоническими синдромами, которые на ранних стадиях могут имитировать болезнь Паркинсона. Длительность наблюдения и тщательная оценка — наиболее важные методы для постановки правильного диагноза.
В табл. 3 представлены основные паркинсонические синдромы, которые необходимо учитывать при проведении дифференциальной диагностики болезни Паркинсона.
/21/21.jpg)
Достаточно часто болезнь Паркинсона дебютирует с неспецифических недвигательных проявлений (боль, депрессия, вегетативные нарушения, расстройства обоняния, эректильная дисфункция и др.), и больные обращаются за помощью к разнообразным специалистам. Первый визит страдающих болезнью Паркинсона к неврологу наблюдается только в 42 % случаев, в 28 % случаев больные обращаются к терапевту, в 6 % — к травматологу, и по 5 % больных впервые обращаются к психиатру, урологу и ревматологу.
Консервативные и нейрохирургические методы лечения болезни Паркинсона и оценка их эффективности. В табл. 4 указаны группы лекарственных препаратов, применяющихся для симптоматического лечения болезни Паркинсона.
Несмотря на то, что болезнь Паркинсона труднокурабельна, большое количество методов лечения целесообразно использовать для лечения ее симптомов и улучшения качества жизни больных. И все же в настоящее время рекомендуются комплексные терапевтические подходы.
Европейская федерация неврологических обществ и Европейский комитет общества экстрапирамидных заболеваний разработали стандарты лечения болезни Паркинсона. Эти стандарты опираются на современные научные познания и практический опыт.
Принятие решения о начале фармакологической терапии у лиц с болезнью Паркинсона должно быть индивидуальным. Задачами первоначальной терапии являются уменьшение двигательных расстройств и улучшение качества жизни больного, не вызывая при этом побочных эффектов.
При решении вопроса о первоначальной терапии и выборе между препаратами леводопы и агонистами допамина врач должен учитывать то, что леводопа намного эффективнее в лечении инвалидизирующих двигательных расстройств у страдающих болезнью Паркинсона, однако при ее применении имеется более высокий риск развития двигательных осложнений (дискинезия и двигательные флуктуации). Агонисты допамина при лечении болезни Паркинсона менее эффективны и вызывают более серьезные нейропсихиатрические осложнения, такие как галлюцинации, спутанность сознания и др., однако при использовании агонистов допамина имеется менее высокий риск развития двигательных осложнений. Двигательные осложнения более характерны для больных молодого возраста (у этих больных первоначальную противопаркинсоническую терапию рекомендуется начинать с агонистов допамина), а психоневрологические осложнения более характерны для больных старшей возрастной группы (у которых первоначальную противопаркинсоническую терапию рекомендуется начинать с препаратов леводопы). У всех больных с когнитивными расстройствами первоначальную терапию следует начинать с леводопы.
По мере прогрессирования заболевания лечение становится более сложным, так как продолжительность действия препаратов леводопы становится короче. Дискинезия становится более выраженной и более продолжительной, вызывая значительную инвалидизацию. Лечение таких больных является искусством, основанным на опыте врача.
В исследованиях препараты леводопы показали себя значительно более эффективными по сравнению со всеми другими группами противопаркинсонических средств (агонисты допамина, ингибиторы МАО-В, антихолинергические препараты и амантадин). Леводопотерапия при болезни Паркинсона ассоциируется с наиболее значительным улучшением двигательных функций, в связи с чем уже после 5 лет лечения подавляющему большинству больных, у которых первоначальная терапия болезни Паркинсона начиналась с агонистов допамина или ингибиторов МАО-В, дополнительно назначают препараты леводопы для лучшего контроля над двигательными паркинсоническими симптомами.
Несмотря на побочные эффекты, ряд исследований показал, что использование препаратов леводопы безопасно даже без специального мониторинга. Важно отметить, что препараты леводопы переносятся лучше, чем любые другие противопаркинсонические средства, особенно у больных старшей возрастной группы.
Одним из основных недостатков препаратов леводопы является короткий период их действия. Обычно терапия препаратами леводопы начинается с их приема 3 раза в день. По мере прогрессирования заболевания, для уменьшения флуктуаций, лучше использовать леводопу небольшими, но частыми дозами или добавить другой противопаркинсонический препарат (например, агонист допамина, ингибитор COMT или ингибитор MAO-B).
Агонисты допамина менее эффективны, чем препараты леводопы, в то же время они могут быть препаратами первого выбора при лечении начальных стадий болезни Паркинсона. Преимуществом агонистов допамина является то, что при их совместном с препаратами леводопы использовании снижается риск развития дискинезии и клинических флуктуаций в первые 5 лет заболевания.
Ингибиторы COMT рекомендуется использовать совместно с леводопой для стабилизации ответа на леводопу, а также для уменьшения клинических флуктуаций и улучшения двигательных функций на поздних стадиях болезни Паркинсона.
Одним из лекарственных средств, используемых при болезни Паркинсона, является амантадин. Изначально амантадин применялся как противовирусный препарат. В 1968 году был впервые обнаружен его противопаркинсонический эффект, и с этого времени амантадин применяется в качестве противопаркинсонического средства.
Данные многочисленных исследований, проведенных за последние 15 лет, указывают на то, что амантадин обладает четким противопаркинсоническим эффектом.
Ингибиторы МАО-В могут быть использованы для симптоматического лечения болезни Паркинсона в качестве монотерапии на начальных стадиях и в комплексе с леводопой на поздних стадиях болезни Паркинсона.
Антихолинергические препараты для лечения болезни Паркинсона в настоящее время используются реже из-за побочных эффектов. В то же время их иногда назначают, если тремор является самым инвалидизирующим симптомом и плохо поддается лечению другими противопаркинсоническими препаратами.
Когда лекарственная терапия становится малоэффективной, должна быть рассмотрена возможность хирургического лечения. Глубокая стимуляция субталамических ядер широко используется для лечения осложненного течения болезни Паркинсона. Результатом нейрохирургического лечения являются уменьшение двигательных симптомов болезни Паркинсона (брадикинезия, тремор и др.), возможность существенного снижения дозировок допаминергических препаратов, уменьшение выраженности леводопа-индуцированной дискинезии и сокращение off-периодов.
По мере прогрессирования заболевания каждый больной нуждается в регулярном наблюдении и модификации схемы лечения.
Использование нефармакологических методов лечения, таких как лечебная физкультура, логопедия и психотерапия, имеет важное значение для оптимизации терапии на поздних стадиях заболевания. Важную роль также играют сестринский осмотр и консультации социальных работников.
Инвалидизация, прогноз. Применение препаратов леводопы, а также других лекарственных средств для лечения двигательных и недвигательных симптомов болезни Паркинсона значительно улучшило качество жизни больных. Но несмотря на то, что имеющиеся лекарственные средства могут значительно уменьшить кардинальные проявления болезни Паркинсона, заболевание неуклонно прогрессирует и его долгосрочный прогноз остается неблагоприятным.
Средняя продолжительность жизни лиц с болезнью Паркинсона составляет от 9,1 до 12,4 года. Через 15 лет после начала болезни более 70 % страдающих болезнью Паркинсона умирают, а половина больных, оставшихся в живых, имеют выраженную инвалидизацию.
Сама по себе болезнь Паркинсона не приводит к смерти. Смерть наступает вследствие сопутствующих заболеваний (бронхолегочные и урологические инфекции, болезни сердца и цереброваскулярные заболевания, злокачественные новообразования и др.) или осложнений (аспирационная пневмония вследствие нарушений глотания, травмы вследствие постуральной неустойчивости и падений и др.).
Атипичный паркинсонизм
Термин «паркинсонизм-плюс» является синонимом термина «атипичный паркинсонизм» и представляет собой группу гетерогенных заболеваний экстрапирамидной системы, проявляющихся паркинсонизмом в сочетании со специфическими дополнительными проявлениями, атипичными для болезни Паркинсона. Атипичный паркинсонизм характеризуется более быстрым прогрессированием по сравнению с болезнью Паркинсона и более короткой продолжительностью жизни больных. К атипичному паркинсонизму относятся мультисистемная атрофия, деменция с тельцами Леви, прогрессирующий надъядерный паралич и кортикобазальная дегенерация.
В настоящее время эти заболевания наиболее удачно классифицируются в соответствии с лежащими в основе патологическими отклонениями и накоплением специфических протеинов на две основные категории: синуклеинопатии и таупатии.
Тау-белок — внутриклеточные включения, обычно выявляющиеся в нейронах с помощью специального окрашивания и состоящие из скрученных или прямых нитевидных структур (нейрофиламентов). Различные по структуре и химическому составу нейрофибриллярные включения (клубочки) могут быть образованы избыточно фосфорилированным тау-белком.
Синуклеинопатии. Синуклеины — семейство белков, обнаруживаемых в нервной ткани и в некоторых видах опухолей. Известно три формы белка: альфа-синуклеин, бета-синуклеин, гамма-синуклеин. Альфа-синуклеин обнаруживается в нервных окончаниях и составляет около одного процента общего белка мозга. Бета-синуклеин в основном экспрессируется в мозге и локализуется в пресинаптических нервных окончаниях. Гамма–синуклеин выявляется преимущественно в периферической нервной системе.
У лиц с БП обнаруживаются цитоплазматические включения, называемые тельцами Леви, основной компонент которых представлен фибриллярной формой белка альфа-синуклеина (А-син). В то же время тельца Леви обнаруживаются в нервных клетках и при других нейродегенеративных заболеваниях, таких как деменция с образованием телец Леви, множественная системная атрофия, фронтотемпоральная дегенерация, на основании чего БП и перечисленные заболевания объединяют в одну нозологическую группу, получившую название «синуклеопатии».
Нейродегенеративные заболевания, обусловленные генетическими изменениями, классифицируются по Mackenzie et al. (2010).
1. Таупатии:
1.1. Болезнь Альцгеймера.
1.2. Прогрессирующий супрануклеарный парез взора.
1.3. Кортикобазальная дегенерация.
1.4. Болезнь серебряного зерна.
1.5. Фронтотемпоральная деменция и паркинсонизм 17-й хромосомы (FTDP–17).
1.6. Болезнь Пика
2. Синуклеопатии:
2.1. Болезнь Паркинсона.
2.2. Деменция с тельцами Леви.
2.3. Мультисистемная атрофия.
2.4. TDP-43 протеинопатия.
2.5. Дегенерация фронтотемпоральных долей с TDP-43 (FTLD-TDP).
Кроме того, выделяют нейродегенеративные заболевания с патологией других белков — фузопатии: дегенерация фронтотемпоральных долей с FUS (FTLD-FUS), Neuronal intermediate filament inclusion disease (NIFID), Basophilic inclusion body disease (BIBD), а также тринуклеотидные и прионные заболевания.
3. Тринуклеотидные заболевания.
3.1. Хорея Хантингтона.
3.2. Спинобульбарная мышечная атрофия, тип Кеннеди.
3.3. Атаксия Фридрейха.
3.4. Спиноцеребеллярная атаксия.
3.5. Дендаторубро-паллидолуизальная атрофия (DRPLA).
4. Прионные заболевания (protein infection, PRION).
4.1. Болезнь Крейтцфельдта — Якоба.
4.2. Синдром Герстмана — Штраусслера — Шейнкера.
4.3. Фатальная семейная бессонница.
4.4. Куру.
Мультисистемная атрофия
Мультисистемная атрофия — прогрессирующее спорадическое нейродегенеративное заболевание неизвестной этиологии, проявляющееся паркинсонизмом в сочетании с различной комбинацией мозжечковых, вегетативных и пирамидных симптомов.
Клинические проявления, особенности течения. Чаще всего мультисистемная атрофия начинается в возрасте 50–55 лет и никогда — в возрасте младше 30 лет.
В зависимости от преобладания тех или иных клинических проявлений выделяют три формы мультисистемной атрофии: паркинсоническую (стриатонигральная дегенерация), мозжечковую (оливопонтоцеребеллярная атрофия) и вегетативную (синдром Шая — Дрейджера).
В большинстве случаев мультисистемная атрофия дебютирует с паркинсонических проявлений и в небольшом числе случаев — с мозжечковой атаксии. Мозжечковые нарушения у больных с мультисистемной атрофией проявляются атаксией при ходьбе и атаксией конечностей, нарушением речи, почерка и нистагмом.
Вегетативные нарушения у больных с мультисистемной атрофией характеризуются ортостатической гипотензией, нарушением потоотделения, расстройствами мочеиспускания (учащенное, задержки, недержание), запорами и редко — недержанием кала. Импотенция у больных с мультисистемной атрофией может развиваться за 5–10 лет до появления других клинических проявлений.
Поведенческие расстройства фазы быстрого сна являются распространенным симптомом мультисистемной атрофии. Обструктивные апноэ во сне также являются частым симптомом мультисистемной атрофии и ассоциируются с внезапной смертью во время сна.
К другим клиническим проявлениям мультисистемной атрофии относятся нарушения глотания и речи, дистония, патологические стопные знаки и гиперрефлексия. К более редким клиническим проявлениям мультисистемной атрофии относят слабость в конечностях, эмоциональную лабильность, миоклонус.
Инструментальные методы диагностики. Дополнительные методы исследования — кардиоваскулярные вегетативные тесты, электромиография анального сфинктера, кардиальная сцинтиграфия, исследование транспортеров допамина, МРТ головного мозга — в некоторых случаях могут быть полезны в диагностике мультисистемной атрофии. Однако большинство исследований с использованием этих диагностических методов проводились на поздних стадиях заболевания, и их диагностическая ценность на ранних стадиях мультисистемной атрофии до настоящего времени неизвестна.
На МРТ у больных с мультисистемной атрофией может обнаруживаться атрофия мозжечка и ствола мозга, а также атрофия и гиподенсивность скорлупы.
Позитронная эмиссионная томография и однофотонная эмиссионная компьютерная томография показывают значительное уменьшение стриатумных допаминергических терминалей у больных с мультисистемной атрофией, однако вышеперечисленные исследования не оказывают помощи в дифференциальной диагностике мультисистемной атрофии с болезнью Паркинсона. Гипометаболизм в скорлупе, стволе мозга и мозжечке может быть отличительным признаком мультисистемной атрофии, выявляемым при проведении позитронно–эмиссионной томографии.
Кардиоваскулярные и другие вегетативные тесты могут выявить различные аспекты вегетативной недостаточности, однако диагностическая ценность этих тестов ограничена.
В настоящее время диагноз мультисистемной атрофии устанавливается на основании новых (2007 г.) диагностических критериев мультисистемной атрофии, которые состоят из 4 частей.
Часть 1. Критерии вероятной мультисистемной атрофии.
Спорадическое прогрессирующее заболевание с дебютом в возрасте старше 30 лет, характеризующееся следующими проявлениями:
— вегетативная дисфункция, проявляющаяся недержанием мочи (неспособность контролировать мочеиспускание, с эректильной дисфункцией у мужчин), или ортостатическое снижение артериального давления в течение 3 минут после вставания как минимум на 30 мм рт.ст. для систолического давления или на 15 мм рт.ст. для диастолического давления и
— плохо поддающийся лечению леводопой паркинсонизм (брадикинезия с ригидностью, тремором или постуральной неустойчивостью) или
— мозжечковый синдром (атаксия при ходьбе с мозжечковой дизартрией, атаксия конечностей или мозжечковые глазодвигательные нарушения).
Часть 2. Критерии возможной мультисистемной атрофии.
Спорадическое прогрессирующее заболевание с дебютом в возрасте старше 30 лет, характеризующееся следующими проявлениями:
— паркинсонизм (брадикинезия с ригидностью, тремором или постуральной неустойчивостью) или
— мозжечковый синдром (атаксия при ходьбе с мозжечковой дизартрией, атаксия конечностей или мозжечковые глазодвигательные нарушения) и
— по крайней мере один признак вегетативной дисфункции, не связанный с каким–либо другим заболеванием (ложные позывы к мочеиспусканию, учащенное мочеиспускание или неполное опорожнение мочевого пузыря, эректильная дисфункция у мужчин или значительная ортостатическая гипотензия, которая не соответствует уровню, необходимому для критериев вероятной мультисистемной атрофии), и
— по крайней мере один дополнительный признак, указанный в части 3.
Часть 3. Дополнительные признаки возможной мультисистемной атрофии.
Возможная мультисистемная атрофия (паркинсоническая или мозжечковая форма):
— положительный рефлекс Бабинского с гиперрефлексией;
— стридор.
Возможная мультисистемная атрофия (паркинсоническая форма):
— быстро прогрессирующий паркинсонизм;
— плохой терапевтический эффект леводопы;
— постуральная неустойчивость, развившаяся в течение 3 лет после появления двигательных симптомов заболевания;
— атаксия при ходьбе, мозжечковая дизартрия, атаксия конечностей или мозжечковые глазодвигательные нарушения;
— дисфагия, развившаяся в течение 5 лет после появления двигательных симптомов заболевания;
— атрофия скорлупы, средних ножек мозжечка, моста или мозжечка на МРТ;
— гипометаболизм в скорлупе, стволе или мозжечке, выявленный при проведении ПЭТ с флюородеоксиглюкозой.
Возможная мультисистемная атрофия (мозжечковая форма):
— паркинсонизм (брадикинезия или ригидность);
— атрофия скорлупы, средних ножек мозжечка или моста на МРТ;
— гипометаболизм в скорлупе, выявленный при проведении ПЭТ с флюородеоксиглюкозой;
— пресинаптическая нигростриальная допаминергическая денервация, выявленная с помощью SPECT и ПЭТ.
Часть 4. Признаки, подтверждающие диагноз мультисистемной атрофии, и признаки, не характерные для мультисистемной атрофии
Признаки, подтверждающие диагноз мультисистемной атрофии:
— орофасциальная дистония;
— диспропорциональный антероколлис;
— камптокормия (выраженный наклон туловища вперед) и/или синдром Пизы (выраженный наклон туловища вбок);
— контрактуры кистей рук или стоп;
— дыхательные нарушения;
— выраженная дисфония;
— выраженная дизартрия;
— недавно появившийся или усилившийся храп;
— холодные ладони и стопы;
— патологический смех или плач;
— толчкообразный, миоклонический постуральный/кинетический тремор.
Признаки, не характерные для мультисистемной атрофии:
— классический тремор покоя по типу «скатывания пилюль»;
— клинически значимая нейропатия;
— не индуцированные лекарствами галлюцинации;
— начало заболевания после 75 лет;
— наследственный анамнез атаксии или паркинсонизма;
— деменция;
— очаги поражения белого вещества, характерные для рассеянного склероза.
До настоящего времени диагноз мультисистемной атрофии основывается на клинических данных.
Дифференциальная диагностика болезни Паркинсона и мультисистемной атрофии часто может вызывать затруднения. Наличие мозжечковых и пирамидных симптомов, выраженные вегетативные расстройства, плохой эффект препаратов леводопы могут служить признаками, отличающими мультисистемную атрофию от болезни Паркинсона. Но эти и ряд других проявлений мультисистемной атрофии могут встречаться и при множестве других неврологических заболеваний, что существенно осложняет диагностику.
Кроме болезни Паркинсона, при проведении дифференциальной диагностики мультисистемной атрофии в первую очередь важно рассматривать другие формы паркинсонизма. Реже мультисистемную атрофию приходится дифференцировать с такими заболеваниями, как спиноцеребеллярные атаксии, позднее начало атаксии Фридрейха, цереброваскулярные заболевания, первично-прогрессирующая форма рассеянного склероза, боковой амиотрофический склероз.
Наиболее распространенными причинами смерти при мультисистемной атрофии являются внезапная смерть во сне неясной этиологии, аспирационная пневмония вследствие нарушений глотания, ортостатическая гипотензия и другие.
Деменция с тельцами Леви
Клинические проявления, особенности течения. Ключевым признаком диагностики деменции с тельцами Леви является прогрессивное ухудшение когнитивных функций, выраженное настолько, что затрудняет выполнение социальных и профессиональных навыков.
К когнитивным расстройствам, отличающим деменцию с тельцами Леви от болезни Альцгеймера, относят более выраженное нарушение внимания и исполнительных функций, а также зрительно-пространственные нарушения. Для деменции с тельцами Леви характерны когнитивные флуктуации, которые могут наблюдаться уже на ранних стадиях заболевания и проявляются эпизодами спутанности сознания и ухудшения интеллектуально-мнестических функций, чередующиеся с нормальными (или близкими к нормальным) состояниями. Наличие когнитивных флуктуаций повышает риск смерти у больных деменцией с тельцами Леви.
Другими психическими расстройствами у больных деменцией с тельцами Леви являются зрительные галлюцинации и галлюцинации в других модальностях (слуховые, тактильные), депрессия и различные нарушения сна (расстройства фазы быстрого сна, чрезмерная дневная сонливость и др.).
Имеются различия паркинсонизма у больных с деменцией с тельцами Леви и с болезнью Паркинсона. При деменции с тельцами Леви наблюдаются более выраженная постуральная неустойчивость и нарушение ходьбы и менее выраженный тремор покоя. Выраженная чувствительность к антипсихотическим препаратам наблюдается у более чем половины лиц с деменцией с тельцами Леви и ассоциируется с более короткой продолжительностью жизни этих больных.
Диагноз деменции с тельцами Леви также подтверждается такими дополнительными признаками, как повторяющиеся падения и синкопе, галлюцинации в других модальностях, зрительно-пространственные расстройства и вегетативные нарушения.
Инструментальные методы диагностики. Нейровизуализационные данные (МРТ и др.) не позволяют подтвердить диагноз деменции с тельцами Леви, но могут оказать помощь при проведении дифференциальной диагностики с сосудистой деменцией и другими заболеваниями.
На МРТ у больных с деменцией с тельцами Леви часто выявляется генерализованная атрофия головного мозга.
При проведении ПЭТ и SPECT определяются уменьшение транспортеров допамина в хвостатом ядре и скорлупе, нигростриальная дегенерация и окципитальная гипоперфузия, что может оказать помощь при проведении дифференциальной диагностики деменции с тельцами Леви и болезни Альцгеймера. У небольшой части больных деменцией с тельцами Леви не отмечается патологических изменений при проведении исследования SPECT.
Диагноз деменции с тельцами Леви устанавливается на основании диагностических критериев заболевания, которые включают центральный признак, основные, наводящие и дополнительные признаки. Также в критериях выделяют симптомы, наличие которых не характерно для деменции с тельцами Леви, а в последнем пункте критериев излагается последовательность развития симптомов при этом заболевании:
Центральный признак:
— прогрессирующая деменция.
Основные признаки:
— когнитивные флуктуации;
— рецидивирующие зрительные галлюцинации;
— паркинсонизм.
Наводящие признаки:
— поведенческие расстройства в фазе быстрого сна;
— выраженная чувствительность к нейролептикам;
— низкий уровень накопления транспортеров допамина в базальных ганглиях, выявленный с помощью SPECT или PET.
Дополнительные признаки:
— повторяющиеся падения и синкопальные состояния;
— транзиторные потери сознания;
— ортостатическая гипотензия, недержание мочи;
— галлюцинации;
— систематизированный бред и мании;
— депрессия;
— зрительно-пространственные расстройства;
— относительная анатомическая сохранность структур височных долей полушарий головного мозга при исследовании с помощью КТ или МРТ;
— генерализованное снижение перфузии на SPECT или ПЭТ со снижением окципитальной активности;
— медленноволновая активность на электроэнцефалограмме с присутствием острых волн в височных отведениях.
Последовательность развития симптомов
Диагноз деменции с тельцами Леви следует устанавливать, когда деменция развилась до паркинсонизма, одновременно с паркинсонизмом или в течение первого года после появления симптомов паркинсонизма.
Деменция с тельцами Леви прогрессирует быстрее, чем болезнь Паркинсона. Средняя продолжительность жизни больных деменцией с тельцами Леви составляет менее 10 лет, у некоторых больных отмечается очень быстрое прогрессирование заболевания и смерть в течение 1–2 лет после появления первых клинических проявлений заболевания.
Прогрессирующий надъядерный паралич
Клинические проявления и особенности течения. Прогрессирующий надъядерный паралич является гетерогенным заболеванием, основными проявлениями которого являются брадикинезия, ригидность, нарушение постуральных рефлексов и падения, паралич взора по вертикали, псевдобульбарный паралич, когнитивные нарушения и другие расстройства.
Одним из самых важных диагностических признаков прогрессирующего надъядерного паралича является паралич взора по вертикали, который может развиваться уже на ранних стадиях заболевания. Сначала развивается паралич взора вниз, а потом — вверх. Горизонтальные движения глазных яблок сохраняются или нарушаются на самых поздних стадиях болезни. У большинства больных с прогрессирующим надъядерным параличом ограничение движений глазных яблок возникает через несколько лет после начала заболевания, а у небольшой части больных не развивается совсем, что существенно осложняет его раннюю диагностику. Ограничение движения глазных яблок вниз считается более специфичным диагностическим признаком прогрессирующего надъядерного паралича.
К другим глазным симптомам, которые могут наблюдаться у больных с прогрессирующим надъядерным параличом, относятся расплывчатость зрения вследствие нарушения конвергенции, блефароспазм и апраксия век.
Падения у больных с прогрессирующим надъядерным параличом могут наблюдаться уже на первом году заболевания.
Деменция у больных с прогрессирующим надъядерным параличом имеет лобно-субкортикальный характер и проявляется забывчивостью, замедленностью мыслительных процессов, изменением личности с апатией или депрессией и нарушением восприятия новой информации. У больных с атипичным течением прогрессирующего надъядерного паралича могут наблюдаться афазия, нарушение праксиса и зрительно–пространственного чувства.
При прогрессирующем надъядерном параличе тремор встречается значительно реже, чем при болезни Паркинсона, и, как правило, он менее выражен.
Для больных с прогрессирующим надъядерным параличом характерна разгибательная поза со слегка наклоненной назад головой, что отличает это заболевание от болезни Паркинсона, для которой характерна сгибательная поза.
При прогрессирующем надъядерном параличе аспирация наблюдается у 30 % больных. Дисфагия коррелирует с длительностью болезни, уровнем инвалидизации и когнитивными нарушениями. Наличие дисфагии на ранних стадиях прогрессирующего надъядерного паралича является предиктором более короткой продолжительности жизни таких больных.
В настоящее время выделяют несколько вариантов прогрессирующего надъядерного паралича, различающихся клинической симптоматикой и патоморфологическими изменениями: классический синдром Ричардсона, ПНП-паркинсонизм, ПНП-неосложненная акинезия с застываниями при ходьбе, ПНП-кортикобазальный синдром, ПНП-афазия и др., что еще более осложняет диагностику этого заболевания.
Инструментальные методы диагностики. На МРТ у больных с прогрессирующим надъядерным параличом могут обнаруживаться атрофия среднего мозга и так называемый симптом «колибри», представляющий собой вогнутую поверхность верхней части среднего мозга (который напоминает колибри) на сагиттальных МРТ-срезах.
При проведении позитронно-эмиссионной томографии и однофотонной эмиссионной компьютерной томографии у больных с прогрессирующим надъядерным параличом обнаруживается снижение метаболизма и перфузии в лобных долях полушарий головного мозга и базальных ганглиях. Исследование метаболизма глюкозы с помощью позитронно-эмиссионной томографии у больных с прогрессирующим надъядерным параличом показывает снижение метаболизма глюкозы в орбитофронтальной области, средней лобной и поясной извилинах, таламусе и среднем мозге по сравнению с контрольной группой здоровых людей того же возраста.
Диагноз прогрессирующего надъядерного паралича устанавливается на основании его диагностических критериев, разработанных Национальным институтом неврологических заболеваний и инсультов (США), а также обществом прогрессирующего надъядерного паралича (the National Institute of Neurological Disorders and Stroke (NINDS) and the Society for Progressive Supranuclear Palsy).
Достоверный диагноз прогрессирующего надъядерного паралича:
— клиническая картина вероятного или возможного прогрессирующего надъядерного паралича плюс гистопатологические доказательства этого заболевания.
Вероятный диагноз прогрессирующего надъядерного паралича:
— постепенно прогрессирующее заболевание с началом в возрасте старше 40 лет;
— надъядерный вертикальный паралич взора и постуральная неустойчивость с падениями в течение первого года болезни;
— отсутствие других заболеваний, объясняющих вышеописанные нарушения (см. критерии исключения).
Возможный диагноз прогрессирующего надъядерного паралича:
— постепенно прогрессирующее заболевание с началом в возрасте старше 40 лет;
— надъядерный вертикальный паралич взора или замедление произвольных движений глазных яблок по вертикали и постуральная неустойчивость с падениями в течение первого года болезни;
— отсутствие других заболеваний, объясняющих указанные выше нарушения (см. критерии исключения).
Критерии исключения для прогрессирующего надъядерного паралича:
— недавно перенесенный энцефалит;
— синдром «чужой руки», корковые чувствительные нарушения, лобная или височно-теменная атрофия;
— галлюцинации или бред, не связанные с допаминергической терапией;
— альцгеймеровский тип корковой деменции (выраженная амнезия и афазия или агнозия);
— ранние мозжечковые симптомы или беспричинная дизавтономия (значительная гипотензия или нарушения мочеиспускания);
— выраженные асимметричные паркинсонические признаки (например, брадикинезия);
— нейровизуализационные данные о наличии структурной патологии (например, инфаркты в области базальных ганглиев или ствола мозга, лобная атрофия);
— болезнь Уиппла, в случае необходимости подтвержденная данными полимеразной цепной реакции.
Подтверждающие критерии ПНП:
— симметричная акинезия или ригидность, более выраженная в проксимальных, чем в дистальных, отделах;
— патологический наклон головы, особенно ретроколлис;
— низкий терапевтический эффект или отсутствие эффекта при лечении паркинсонизма препаратами леводопы;
— раннее развитие дизартрии и дисфагии;
— раннее развитие когнитивных нарушений, включающих по крайней мере два из перечисленных признаков: апатия, нарушения абстрактного мышления, снижение беглости речи, нарушения поведения или лобные симптомы.
Инвалидизация, прогноз. Уже через 3 года после начала заболевания половина больных с прогрессирующим надъядерным параличом не может передвигаться без посторонней помощи.
Основной причиной смерти больных с прогрессирующим надъядерным параличом является развившаяся вследствие нарушения глотания аспирационная пневмония.
Кортикобазальная дегенерация
Кортикобазальная дегенерация — нейродегенеративное заболевание, характеризующееся атрофией, глиозом и тау-иммунореактивными патологическими изменениями в сером и белом веществе неокортекса, базальных ганглиях и черной субстанции.
Клиническая картина. Средний возраст дебюта кортикобазальной дегенерации составляет 63 года. Клиническая картина кортикобазальной дегенерации проявляется асимметричным паркинсонизмом, идеомоторной апраксией руки, дистонией, миоклонусом, нарушением равновесия и ходьбы, лобной деменцией, изменением речи (дизартрия, афония) и другими психическими нарушениями (апатия, депрессия, поведенческие и личностные изменения). Кроме этого, у больных с кортикобазальной дегенерацией иногда могут наблюдаться гиперрефлексия, патологические стопные и субкортикальные рефлексы и мозжечковые знаки.
Наиболее распространенными паркинсоническими проявлениями кортикобазальной дегенерации являются ригидность (92 % случаев), брадикинезия и нарушения ходьбы (по 80 % случаев). Дистония руки наблюдается у 59 % больных с кортикобазальной дегенерацией в отличие от дистонии туловища или ног, которая при этом заболевании встречается довольно редко.
Тремор покоя у больных с кортикобазальной дегенерацией наблюдается в 29 % случаев, что еще больше затрудняет дифференциальную диагностику с болезнью Паркинсона.
Специфическим проявлением кортикобазальной дегенерации является симптом «чужой руки» (alien hand phenomenon), который наблюдается в 60 % случаев этого заболевания. Симптом «чужой руки» проявляется неспособностью осознавать и контролировать действия одной из рук, которая не подчиняется произвольному контролю. Симптом «чужой руки» достаточно хорошо представлен в фильме о Гарри Поттере, где одному из персонажей приходится бороться с собственной рукой, которая хочет его задушить.
И все же наиболее часто первыми проявлениями кортикобазальной дегенерации являются неловкость в руке (50 % случаев), нарушения ходьбы (36 %), односторонняя болезненная парестезия (29 %), лобная деменция (21 %), падения (21 %), дизартрия (14 %), депрессия (7 %).
Когнитивные нарушения в начале заболевания менее заметны, чем двигательные, но позже нарастают по мере прогрессирования заболевания. Деменция у больных с кортикобазальной дегенерацией может как присутствовать в самом начале заболевания, так и вовсе отсутствовать даже на его поздних стадиях.
Описаны атипичные случаи кортикобазальной дегенерации с симметричным началом и отсутствием таких типичных признаков данного заболевания, как синдром «чужой руки» и дистония руки, что еще более усложняет его диагностику.
Диагноз, дифференциальная диагностика, диагностические критерии. Диагностические ошибки при постановке диагноза кортикобазальной дегенерации случаются очень часто, несмотря на наличие некоторых характерных признаков данного заболевания. Диагностическая точность при первом визите составляет только 35 %.
Многим больным с кортикобазальной дегенерацией ошибочно выставляются такие диагнозы, как болезнь Паркинсона, прогрессирующий надъядерный паралич, мультисистемная атрофия, и диагнозы других нейродегенеративных заболеваний.
Низкая доля больных с кортикобазальной дегенерацией с правильно поставленным диагнозом может быть обусловлена плохой осведомленностью неврологов об этом заболевании.
Существуют разнообразные диагностические критерии диагностики кортикобазальной дегенерации и кортикобазального синдрома (Кембриджские критерии, критерии Торонто и критерии Майо) (2013).
Клинические критерии вероятной спорадической кортикобазальной дегенерации:
— постепенное начало и прогрессирование;
— наличие симптомов не менее одного года;
— возраст начала заболевания старше 50 лет;
— отсутствие семейного анамнеза (двое и более родственников);
— возможные фенотипы (см. клинические синдромы): 1) вероятный кортикобазальный синдром или 2) лобный синдром с поведенческими и пространственными нарушениями или первично-прогрессирующая афазия плюс по крайней мере один признак кортикобазального синдрома;
— отсутствие генетической мутации, связанной с тау.
Клинические критерии возможной кортикобазальной дегенерации:
— постепенное начало и прогрессирование;
— наличие симптомов в течение не менее одного года;
— возможные фенотипы (см. клинические синдромы): 1) вероятный кортикобазальный синдром, или 2) лобный синдром с поведенческими и пространственными нарушениями или первично-прогрессирующая афазия, или 3) синдром прогрессирующего надъядерного паралича плюс по крайней мере один признак кортикобазального синдрома.
Клинические фенотипы (синдромы), связанные с кортикобазальной дегенерацией:
— вероятный кортикобазальный синдром. Асимметричное течение с наличием двух из следующих признаков: a) ригидность и акинезия конечностей; б) дистония конечностей; в) миоклонус конечностей и двух из следующих признаков: а) апраксия конечностей, или оробуккальная апраксия; б) корковые чувствительные нарушения; в) синдром «чужой руки» (alien limb phenomenon);
— возможный кортикобазальный синдром. Может быть симметричным. Наличие одного из следующих признаков: a) ригидность и акинезия конечностей; б) дистония конечностей; в) миоклонус конечностей и одного из следующих признаков: а) апраксия конечностей или оробуккальная апраксия; б) корковые чувствительные нарушения; в) синдром «чужой руки»;
— лобный синдром с поведенческими и пространственными нарушениями. Наличие двух из следующих признаков: a) нарушения исполнительных функций; б) поведенческие или личностные расстройства; в) зрительно-пространственный дефицит;
— первично-прогрессирующая афазия (аграмматическая форма). Затрудненная, аграмматическая речь плюс наличие по крайней мере одного из следующих признаков: a) нарушения понимания предложений при относительно сохранившемся понимании отдельных слов; б) апраксия речи;
— синдром прогрессирующего надъядерного паралича. Наличие трех из следующих признаков: a) аксиальная ригидность, или симметричная ригидность конечностей, или акинезия; б) постуральная неустойчивость или падения; в) недержание мочи; г) поведенческие нарушения; д) надъядерный вертикальный паралич взора или уменьшение амплитуды произвольных вертикальных движений глазных яблок.
Критерии исключения кортикобазальной дегенерации:
1) наличие признаков заболевания, связанного с тельцами Леви: классический паркинсонический тремор покоя, отличный и продолжительный эффект леводопы, наличие галлюцинаций;
2) наличие признаков мультисистемной атрофии: дизавтономия или явные мозжечковые нарушения;
3) наличие признаков бокового амиотрофического склероза: признаки поражения как верхнего, так и нижнего мотонейрона;
4) семантический или логопедический вариант первично-прогрессирующей афазии;
5) структурные поражения, указывающие на очаговую причину заболевания;
6) гранулиновая мутация или пониженный уровень програнулина в плазме, мутации TDP-43 или FUS;
7) наличие признаков болезни Альцгеймера.
На МРТ у больных с кортикобазальной дегенерацией определяются асимметричная атрофия коры лобно-теменной области, атрофия средней части мозолистого тела, скорлупы, увеличение латерального и третьего желудочков. Атрофические изменения на МРТ у больных с кортикобазальной дегенерацией более выражены, чем у больных с другими нейродегенеративными заболеваниями.
Исследование метаболизма глюкозы с помощью позитронно-эмиссионной томографии у больных с кортикобазальной дегенерацией показало асимметрию метаболизма глюкозы в теменной и лобной долях головного мозга, а также поясной извилине.
Прогноз
Неизбежным исходом кортикобазальной дегенерации является выраженная инвалидизация.
Средняя продолжительность жизни больных с кортикобазальной дегенерацией от начала первых клинических симптомов составляет 7,9 года (от 2,5 до 12,5 года). Низкая продолжительность жизни обусловлена двухсторонним паркинсонизмом и лобной деменцией. Бронхопневмония как результат дисфагии и обездвиженности является непосредственной причиной смерти большинства больных.
Сосудистый паркинсонизм
Цереброваскулярные заболевания являются не таким частым фактором паркинсонизма и составляют 12 % от всех его случаев.
Клинические проявления. Первая форма (самая распространенная) встречается в основном у больных с анамнезом артериальной гипертензии и проявляется нарушением ходьбы, симметричной ригидностью, отсутствием тремора. Вторая форма отличается более постепенным началом паркинсонизма, сходством с идиопатической болезнью Паркинсона и небольшой эффективностью допаминергических препаратов. Третья форма (наиболее редкая) проявляется внезапно возникающим паркинсонизмом вследствие сосудистого поражения базальных ганглиев.
У больных с сосудистым паркинсонизмом отмечаются более выраженные шаркающая походка, застывания при ходьбе, постуральная неустойчивость и чаще наблюдаются пирамидные симптомы, псевдобульбарный паралич, недержание мочи и когнитивные нарушения по сравнению с лицами с болезнью Паркинсона. В то же время тремор и паркинсонические симптомы в верхних конечностях у больных с сосудистым паркинсонизмом выражены в меньшей степени, чем у страдающих болезнью Паркинсона. Хоботковый и ладонно–подбородочный рефлексы также могут быть дифференциальными признаками, отличающими сосудистый паркинсонизм от болезни Паркинсона.
Возникновение сосудистого паркинсонизма сразу после инсульта или в течение одного года после инсульта наблюдается у 25 % больных.
Диагностические критерии. В настоящее время диагностика сосудистого паркинсонизма довольно часто вызывает затруднения у специалистов. В первую очередь потому, что на данный момент не существует широко распространенных диагностических критериев сосудистого паркинсонизма.
Для диагностики сосудистого паркинсонизма в настоящее время могут быть использованы диагностические критерии, предложенные J.C.M. Zijlmans и соавт. в 2004 г.:
А. Паркинсонизм: брадикинезия и по крайней мере один из перечисленных ниже синдромов: тремор покоя, мышечная ригидность или постуральная неустойчивость, не связанная с первичной зрительной, вестибулярной, мозжечковой или проприоцептивной дисфункцией.
Б. Цереброваскулярное заболевание, подтвержденное данными КТ или МРТ, или наличие очаговых признаков или симптомов, указывающих на инсульт.
В. Наличие связи между этими двумя заболеваниями: 1) острое или отсроченное начало на фоне инфарктов в области черной субстанции (pars compacta), или вентролатеральных ядер таламуса, или обширного инфаркта лобной доли, начало паркинсонизма с контралатерального акинетико-ригидного синдрома или шаркающей походки в течение одного года после инсульта; 2) постепенное начало паркинсонизма с обширным поражением субкортикального белого вещества, двухстороннее начало паркинсонизма и наличие шаркающей походки на начальных стадиях заболевания или ранние когнитивные нарушения.
Критерии исключения сосудистого паркинсонизма:
— повторные черепно-мозговые травмы в анамнезе;
— дебют заболевания на фоне лечения нейролептиками;
— наличие опухоли головного мозга или сообщающейся гидроцефалии, подтвержденных данными КТ или МРТ, или другие альтернативные объяснения паркинсонизма.
Инструментальные методы диагностики и дифференциальная диагностика
Цереброваскулярные факторы риска (артериальная гипертензия, повышенный уровень холестерина) чаще встречаются у больных с сосудистым паркинсонизмом по сравнению со страдающими болезнью Паркинсона.
Практически у всех больных с сосудистым паркинсонизмом на МРТ имеются патологические ишемические изменения в области базальных ганглиев, субкортикальном и перивентрикулярном белом веществе головного мозга. Атрофические изменения головного мозга также могут часто наблюдаться при проведении МРТ. Имеется корреляция между атрофией лобных долей головного мозга и нарушением ходьбы у пациентов с сосудистым паркинсонизмом, но в то же время не имеется специфического МРТ-паттерна, характерного для таких больных. Не наблюдается достоверных нейровизуализационных МРТ-различий между группой больных с сосудистым паркинсонизмом и лицами с болезнью Бинсвангера. В то же время лакунарные инфаркты в области базальных ганглиев могут быть обнаружены у больных без клинических проявлений паркинсонизма и даже у пациентов без какой-либо неврологической симптоматики.
При проведении дифференциальной диагностики сосудистого паркинсонизма может быть полезна однофотонная эмиссионная компьютерная томография. У пациентов с сосудистым паркинсонизмом и болезнью Паркинсона на DAT SPECT отмечается значительный пресинаптический допаминергический дефицит по сравнению с контрольной группой здоровых лиц. Индекс асимметрии пресинаптических допаминергических изменений у больных с сосудистым паркинсонизмом был таким же, как и у здоровых лиц, но ниже, чем у страдающих болезнью Паркинсона. Пресинаптический допаминергический дефицит у больных с сосудистым паркинсонизмом ассоциировался с тяжестью заболевания и не ассоциировался с его длительностью.
Тем не менее довольно сложно рассуждать о роли томографов DAT в дифференциальной диагностике сосудистого паркинсонизма. Отсутствие унифицированных клинических критериев сосудистого паркинсонизма приводит к трудностям интерпретации результатов. В ряде случаев полученные результаты противоречат друг другу.
В заключение лекции следует отметить, что дебют паркинсонических синдромов все же приходится на более поздний возрастной период по сравнению с дебютом болезни Паркинсона. Атипичные варианты паркинсонизма (мультисистемная атрофия, деменция с тельцами Леви, прогрессирующий надъядерный паралич, кортикобазальная дегенерация), а также сосудистый паркинсонизм отличаются более быстрым прогрессированием, неблагоприятным течением по сравнению с болезнью Паркинсона, наличием ранних нарушений ходьбы и равновесия, галлюцинаций и спутанности сознания, а также выраженных когнитивных и вегетативных нарушений. Среди паркинсонических синдромов наиболее неблагоприятное течение имеют кортикобазальная дегенерация, деменция с тельцами Леви и прогрессирующий надъядерный паралич. Уже в первые 3 года болезни эти больные имеют умеренную или выраженную инвалидизацию и зависят от окружающих при самообслуживании. Мультисистемная атрофия и сосудистый паркинсонизм имеют более прогредиентное течение по сравнению с болезнью Паркинсона, но менее прогредиентное по сравнению с кортикобазальной дегенерацией, деменцией с тельцами Леви и прогрессирующим надъядерным параличом. При болезни Паркинсона до 80 % пациентов имеют положительный эффект от допаминергических препаратов (леводопа, агонисты допамина), а при паркинсонических синдромах допаминергические препараты менее эффективны.
1. Паркинсон Дж. Эссе о дрожательном параличе: Пер. с англ. проф. М.В. Селиханова. — СПб., 2010. — 80 с.
2. Влияние немоторных нарушений на качество жизни больных болезнью Паркинсона / Т.Н. Калищук-Слободин, Ю.И. Головченко, С.И. Шкробот и др. // Український вісник психоневрології. — 2007. — Т. 15, вип. 1 (додаток). — С. 58–59.
3. Карабань Н.В. Применение леводопасодержащих препаратов на современном этапе лечения болезни Паркинсона / Н.В. Карабань // Міжнародний неврологічний журнал. — 2006. — № 6. — С. 16–20.
4. Мультисистемная атрофия мозга как наиболее вероятный диагноз у больного шестидесяти лет с паркинсоническим синдромом десятилетней давности / С.К. Евтушенко, Р.В. Симанов // Міжнародний неврологічний журнал. — 2012. — № 5. — С. 86–90.
5. Суховерская О. Болезнь Паркинсона и паркинсонические синдромы: диагноз и лечение / О. Суховерская // Міжнародний неврологічний журнал. — 2011. — № 6. — С. 16–24.
6. Труфанов Е.А. Дифференциальный диагноз и прогноз болезни Паркинсона, паркинсонических синдромов и эссенциального тремора: автореф. на здобуття наук. ступеня д. мед. наук: спец. 14.01.15 «Неврологія» / Е.А. Труфанов. — Луганск, 2013. — 464 с.
7. (123I)FP-CIT SPECT in suspected Dementia with Lewy Bodies: a longitudinal case study / F.J. Siepel, A. Rongve, T.C. Buter et al. // BMJ Open. — 2013. — Vol. 3, № 4. — P. 1–8.
8. [123I]FP-CIT Spect study in Vascular Parkinsonism and Parkinson’s Disease / J. Zijlmans, A. Evans, F. Fontes et al. // Movement Disorders. — 2007. — Vol. 2, № 9. — P. 1278–1285.
9. A new MRI rating scale for Progressive Supranuclear Palsy and Multiple System Atrophy: validity and reliability / Y. Rolland, M. Verin, C.A. Payan et al. // Journal of Neurology, Neurosurgery & Psychiatry. — 2011. — Vol. 82, № 9. — P. 1025–1032.
10. A validation exercise on the new consensus criteria for Multiple System Atrophy / Y. Osaki, Y. Ben-Shlomo, A.J. Lees et al. // Movement Disorders. — 2009. — Vol. 24, № 15. — P. 2272–2276.
11. Bogaerts V. Genetic findings in Parkinson’s Disease and translation into treatment: a leading role for mitochondria? / V. Bogaerts, J. Theuns, C. van Broeckhoven // Genes, Brain and Behavior. — 2008. — Vol. 7, № 2. — P. 129–151.
12. Clinicopathological investigation of Vascular Parkinsonism, including clinical criteria for diagnosis / J.C.M. Zijlmans, S.E. Daniel, A.J. Hughes et al. // Movement Disorders. — 2004. — Vol. 19, № 6. — P. 630–640.
13. Determinants of the timing of symptomatic treatment in early Parkinson Disease: the National Institutes of Health Exploratory Trials in Parkinson Disease (NET-PD) Experience / S.A. Parashos, C.J. Swearingen, K.M. Biglan et al. // Archives of Neurology. — 2009. — Vol. 66, № 9. — P. 1099–1104.
14. Diagnosis and management of Dementia with Lewy Bodies. Third Report of the DLB Consortium / I.G. McKeith, D.W. Dickson, J. Lowe et al. // Neurology. — 2005. — Vol. 65, № 12. — P. 1863–1872.
15. Epidemiology of Multiple System Atrophy. ESGAP Consortium. European Study Group on Atypical Parkinsonisms / N. Vanacore, V. Bonifati, G. Fabbrini et al. // Neurological Sciences. — 2001. — Vol. 22, № 1. — P. 97–99.
16. Fahs S. Parkinsons Disease: 10 years of progress, 1997–2007 / S. Fahn // Movement Disorders. — 2010. — Vol. 25 (Suppl. 1). — P. S2–S14.
17. Genetic etiology of Parkinson Disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: a mutation update / K. Nuytemans, J. Theuns, M. Cruts, C. Van Broeckhoven // Human Mutation. — 2010. — Vol. 31, № 7. — P. 763–780.
18. Gilman S. Parkinsonian syndromes / S. Gilman // Clinics in Geriatric Medicine. — 2006. — Vol. 22, № 4. — P. 827–842.
19. Graeber M.B. Biomarkers for Parkinson’s Disease / M.B. Graeber // Experimental Neurology. — 2009. — Vol. 216, № 2. — P. 249–253.
20. Jankovic J. Parkinson’s Disease: clinical features and diagnosis / J. Jankovic // Journal of Neurology, Neurosurgery & Psychiatry. — 2008. — Vol. 79, № 4. — P. 368–376.