Вступ
Кінцівково-поясна м’язова дистрофія (КПМД) представляє групу гетерогенних генетичних порушень з різним віком початку, що в першу чергу викликають слабкість проксимальних м’язів плечового і тазового пояса.
Типовими клінічними проявами цієї групи захворювань є порушення ходи (качина хода), прийом Говерса (підйом драбинкою з положення навпочіпки), гіперлордоз у поперековому відділі хребта, крилоподібні лопатки, симптом млявих надпліч, м’язова гіпотонія і гіпотрофія, сухожилкова гіпорефлексія, що пропорційна м’язовій слабкості. При біохімічному дослідженні спостерігається збільшення рівня активності ферменту креатинфосфокінази у плазмі крові.
Для усіх виявлених варіантів КПМД проведено картування гена на певній хромосомі, для деяких із них ідентифіковані основні типи патологічних мутацій і білковий продукт експресії. На сьогодні КПМД має більше 30 різних підтипів, пов’язаних із специфічними генними локусами, що проявляються перехрещеними та гетерогенними фенотипами [1–3, 6].
Кінцівково-поясна м’язова дистрофія класифікована на основі типу успадкування на 2 основні типи: автосомно-домінантний (AД-КПМД1) і автосомно-рецесивний (AР-КПМД2) [4]. До кожного типу додається англійська літера абетки в хронологічному порядку, кожен раз, коли повідомляється про новий хромосомний локус, і далі зазначають уражений білок. Так схематично виглядає формулювання діагнозу: «КПМД, успадкування (Р або Д), порядок виявлення (число), уражений білок».
Станом на 2018 рік відомо про існування 8 підтипів КПМД типу 1 і 26 підтипів КПМД типу 2 [5] залежно від дефектного білка саркогліканового та дистрогліканового комплексів [6] (рис. 1) [7]. Х-зчеплені розлади, що проявляються патерном слабкості проксимальних м’язів плечового і тазового поясу та включають м’язову дистрофію Емері — Дрейфуса і дистрофінопатії (м’язова дистрофія Дюшена, м’язова дистрофія Беккера та носії дистрофінопатій), не включені до категорії КПМД.
З огляду на генетичну і фенотипову гетерогенність даної патології, КПМД слід розглядати практично в усіх пацієнтів, які звертаються зі скаргами на первинну м’язову слабкість.
/22.jpg)
За різними оцінками, загальна поширеність синдромів КПМД становить 1,63 на 100 000 (від 0,56 до 5,75) [6, 8] та залежить від підтипу та географії. Це четверта за частотою м’язова дистрофія після дистрофінопатій, міотонічної дистрофії та лице-плечелопаткової дистрофії. Основна кількість КПМД успадковується за автосомно-рецесивним типом, і лише близько 10 % припадає на автосомно-домінантні форми. Автосомно-домінантні та автосомно-рецесивні форми КПМД з однаковою частотою зустрічаються в осіб обох статей. Вік початку варіює серед різних мутацій та становить від 1 до 50 років, хоча у деяких пацієнтів КПМД може бути безсимптомною. Це також може варіювати серед сімей та членів родини з тією ж мутацією. Взагалі пацієнти з автосомно-домінантною КМПД мають більш пізній початок і більш повільний перебіг захворювання, ніж хворі з автосомно-рецесивним типом КМПД. Підвищення креатинкінази (КФК) також є менш значущим при автосомно-домінантній КПМД, ніж при автосомно-рецесивних формах.
Диференціальна діагностика має проводитися при низці захворювань, враховуючи насамперед ознаки прогресуючої симетричної проксимальної м’язової слабкості кінцівок, що характеризуються розвитком аналогічних симптомів у різних комбінаціях: колаген- VI-пов’язані міопатії (Бетлема та Ульріха), ендокринні та метаболічні міопатії, хвороби накопичення (зокрема, хвороба Помпе), прогресуюча м’язова дистрофія, дистрофінопатія Беккера, тазово-стегнова міодистрофія Лейдена — Мебіуса, лице-плечелопаткова м’язова дистрофія, спінальна м’язова атрофія та запальні міопатії (дермато- й поліміозити).
З огляду на генетичну та фенотипову гетерогенність даної патології, КПМД слід розглядати практично у всіх пацієнтів, які звертаються зі скаргами на первинну м’язову слабкість.
На сьогодні список генів для скринінгу занадто великий і більш підходить для цільових панелей секвестрування наступного покоління (NGS), що повинні включати будь-який ген, який до цього часу асоціювався з клінічною картиною КПМД. Магнітно-резонансна томографія (МРТ) допомагає диференціювати окремі форми та підтипи КПМД. Зміна гіперінтенсивного сигналу при скануванні на Т1-ЗЗ спостерігається в уражених м’язах. МРТ-дослідження показало, що залежно від типу та підтипу КПМД відзначається ураження окремих груп м’язів нижніх кінцівок. Наприклад, пацієнти з КПМД типу 2A демонструють помітне залучення аддукторів стегна, підколінних сухожилків і медіальної головки литкового м’яза, водночас збережений кравецький м’яз, крім того, візуалізація показує залучення підошовного м’яза, медіального широкого та двоголового м’яза стегна [9]. Пацієнти з КПМД типу 2B можуть мати змінну картину МРТ, в основному задіяні привідні м’язи, чотириголовий м’яз і м’язи гомілки з відносним збереженням кравцевого і тонкого м’язів [10]. Участь литкового м’яза переважає при міопатії Міоші, ураження сідничних м’язів і передньої та задньої груп м’язів стегна є більш характерними для пацієнтів з фенотипом КПМД. Пацієнти з саркогліканопатією не демонструють будь-яких істотних розбіжностей у характері ураження м’язів на МРТ. Привідні м’язи та сідничні, ймовірніше, уражаються першими, що проявляється ураженням латерального широкого м’яза стегна. Довгий привідний м’яз може мати деякі ділянки збереження з повним або відносним збереженням заднього великогомілкового м’яза та довгого згинача пальців. Також ми можемо бачити відносну гіпертрофію правцевого та тонкого м’яза [11]. Пацієнти з КПМД типу 2D і м’язовою дистрофією Беккера мали більш серйозні МРТ-зміни передньої групи м’язів стегна, ніж задньої. М’язова тканина у пацієнтів з КПМД типу 2I, які мають мутацію FKRP, демонструє характерний патерн у вигляді жирової інфільтрації і набряку при МРТ-дослідженні проміжного широкого та медіального широкого м’язів [12].
На останньому, 229-му міжнародному семінарі Європейського нервово-м’язового центру (ENMC), м. Наарден, Нідерланди, що відбувся 17–19 березня 2017 р. і був присвячений кінцівково-поясній м’язовій дистрофії, були розглянуті та уточнені питання номенклатури і класифікації останньої [13]. Був досягнутий консенсус щодо збереження терміну «кінцівково-поясна м’язова дистрофія», а також уточнено саме визначення терміну. Деякі ознаки були визначені як важливі для виключення інших станів, таких як вроджена м’язова дистрофія і деякі міопатії.
Поряд зі стандартними методами діагностики м’язової патології, що давно увійшли до практики лікаря-невролога (електроміографія, біопсія), визначена роль МРТ для виявлення або оцінки ступеня дистрофічних змін у м’язах, які були визначені як заміна скелетних м’язів жировою тканиною, виявлена на стандартних T1-зважених осьових зображеннях. Протягом останніх десятиліть магнітно-резонансна томографія стала вирішальною для діагностики захворювань м’яких тканин. Її цінність була переконливо доведена при спадкових і запальних нервово-м’язових захворюваннях. Поряд з неврологічним і нейрофізіологічним дослідженням м’язова МРТ є цінним діагностичним інструментом, що дозволяє оцінити ступінь і, що більш важливо, звузити діагностичний пошук у випадках складнокурабельних, передусім м’язових, захворювань [14–17]. МРТ м’язів нижніх кінцівок, як було доведено, є надійним неінвазивним інструментом для діагностики й оцінки прогресування нервово-м’язових захворювань, що демонструє специфічні патерни ураження м’язів нижніх кінцівок при безлічі міопатій [18, 19].
Клінічний випадок
Наводимо клінічний випадок пацієнтки, яка перебуває під спостереженням в Обласному клінічному центрі нейрохірургії та неврології м. Ужгород, у діагностиці якої були використані сучасні методи діагностики, зокрема МРТ м’язів та генетичне консультування, що дало нам змогу скоротити шлях до правильного діагностичного рішення.
Хвора З., 31 рік. Перебуває під спостереженням нашого центру протягом 2,5 року. Звернулась із скаргами на слабкість та відчуття стягування в ногах (стегна), порушення ходи, труднощі при вставанні зі стільця та підйомі по сходинках.
Із анамнезу: вважає себе хворою протягом 12 років, коли стала відчувати слабкість у ногах. Поступово слабкість зростала, з’явились болі в нижніх кінцівках, стало важко ходити (останні 5–7 років), вставати зі стільця, з’явились труднощі при підйомі по сходинках, піднятті рук догори (під час розчісування), в останні 3 роки змінилася хода.
Сімейний анамнез: без спадкової патології.
Неврологічний статус: власна мова чітка. Обличчя симетричне, язик розташований медіально. Ковтання не порушено. Глотковий рефлекс збережений. Рухливість м’якого піднебіння при фонації збережена. Сухожилкові рефлекси з рук збережені, колінні abs, ахілові з клонусами стоп. Гіпотонія м’язів нижніх кінцівок. Атрофії/гіпотрофії не виявлено (гомілка: права та ліва — 33 см, стегна: праве та ліве — 44 см). М’язова сила в нижніх кінцівках до 3,0 бала (проксимально), в руках — до 4,0 бала (проксимально). Чутливих розладів не показує. Стояння на носках збережене, на п’ятах — утруднене. Позитивний симптом Говерса. Хода качина. MRS — 4 бали, MDS — 31 бал, шкала Віньоса — 3.
КФК 929,0. ALT 52,0. AST 47,0. Rö-ОГК, ЕхоКС, спірометрія, спірографія, кардіолог — норма. Генетичний аналіз альфа-галактозидази негативний. МРТ поперекового відділу хребта: гіперлордоз, периневральна кіста Тарлова рівня S2.
Пацієнтка консультована ревматологом, виключені запальні міопатії — поліміозит і дерматоміозит.
ЕНМГ: по моторним та сенсорним волокнам верхніх і нижніх кінцівок — норма; при тестуванні нервово-м’язової передачі — блокування по міастенічному типу не виявлено; ЕМГ-ознаки генералізованого, міопатичного типу, ураження з перевагою у нижніх кінцівках більш проксимально.
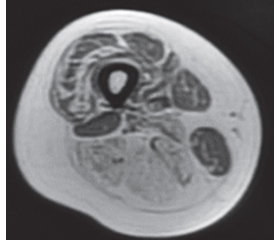
Нами було виконано МРТ м’язів тазового пояса (сканер GE, Signa HDxt 1.5T, ЗДЦ «ЄвроКлінік», м. Ужгород) із застосуванням Т1-, Т2- та STIR-послідовностей: інтактність з обох боків: m.rectus femoris, m.sartorius, m.gracilis, m.biceps femoris: ccaput brevis; двобічне субтотальне заміщення жировою тканиною волокон м’язів стегон передньої та задньої групи, крім того, атрофічні зміни з заміщенням жировою тканиною м’язів гомілок задньої групи двобічно, 1-ша — 3-тя стадія за шкалами Mercuri та Fischer (рис. 2, 3).
Консультована генетиком, виконана NGS-панель (табл. 1). Виявлена мутація у гені CAPN3 у гомозиготному стані. Ген CAPN3 (кальпокаїн 3) пов’язаний з автосомно-рецесивним типом КПМД типу 2А.
На підставі всіх проведених клінічних та інструментальних методів обстеження, з огляду в тому числі й на клінічну картину, пацієнтці було встановлено діагноз: КПМД типу 2А, CAPN3 з тетрапарезом (легким у руках, помірним у ногах), порушеннями функції ходи.
Висновки
Таким чином, враховуючи наш досвід, пацієнт з міопатичним синдромом потребує комплексного клініко-діагностичного підходу із залученням суміжних спеціалістів. Необхідно проводити повне клініко-біохімічне обстеження із визначенням у тому числі рівня КФК, АЛТ, АСТ. Результати лабораторних обстежень потребують ретельного зіставлення з даними клінічного огляду та результатами інструментальних методів обстеження, зокрема з даними ЕНМГ та МРТ м’язів. Клінічне застосування МРТ м’язів та ЕНМГ допомагає у ранній діагностиці прогресуючих форм м’язових дистрофій, що в подальшому дозволить своєчасно призначити лікування та визначити реабілітаційні заходи.
Конфлікт інтересів. Автори заявляють про відсутність конфлікту інтересів при підготовці цієї статті.
Список литературы
1. Nigro V., Savarese M. Genetic basis of limb-girdle muscular dystrophies: the 2014 update. Acta Myol. 2014. № 33. Р. 1-12.
2. Fanin M., Angelini C. Progress and challenges in diagnosis of dysferlinopathy. Muscle Nerve. 2016. № 54. Р. 821-835.
3. Bushby K.M. The limb-girdle muscular dystrophies-multiple genes, multiple mechanisms. Hum. Mol. Genet. 1999. № 8. Р. 1875-1882.
4. Bushby K.M., Beckmann J.S. The limb-girdle muscular dystrophies proposal for a new nomenclature. Neuromuscul. Disord. 1995. № 5(4). Р. 337-435.
5. Chu M.L., Moran E. The Limb-Girdle Muscular Dystrophies: Is Treatment on the Horizon? Neurotherapeutics. 2018. № 15. Р. 849-862.
6. Евтушенко С.К., Шаймурзин М.Р., Евтушенко О.С., Евтушенко И.С. Нейромышечные заболевания у детей. Монография. Донецк: Ноулидж (донецкое отделение), 2014. 218 с.
7. Гришина Д.А., Супонева Н.А., Шведков В.В., Белопасова А.В. Наследственная прогрессирующая конечностно-поясная мышечная дистрофия 2А типа (кальпаинопатия): обзор литературы. Нервно-мышечные болезни. 2015. № 1. С. 25-36.
8. Liewluck T., Milone M. Untangling the complexity of limb-girdle muscular dystrophies. Muscle&Nerve. 2018. № 58(2). Р. 167-177.
9. Richard I., Hogrel J.Y., Stockholm D., Payan C.A., Fougerousse F. et al., Calpainopathy Study Group. Natural history of LGMD2A for delineating outcome measures in clinical trials. Ann. Clin. Transl. Neurol. 2016 Apr. № 3(4). Р. 248-65.
10. Jin S., Du J., Wang Z., Zhang W., Lv H., Meng L. et al. Heterogeneous characteristics of MRI changes of thigh muscles in patients with dysferlinopathy. Muscle Nerve. 2016 Dec. № 54(6). Р. 1072-1079.
11. Tasca G., Monforte M., Díaz-Manera J. et al. MRI in sarcoglycanopathies: a large international cohort study. J. Neurol. Neurosurg. Psychiatry. 2018 Jan. № 89(1). Р. 72-77.
12. Xie Z., Xiao J., Zheng Y., Wang Z., Yuan Y. Magnetic Resonance Imaging Findings in the Muscle Tissue of Patients with Limb Girdle Muscular Dystrophy Type 2I Harboring the Founder Mutation c.545A>G in the FKRP Gene. Biomed. Res. Int. 2018. № 2018. 3710814.
13. Straub V., Corrado A. et al. 229th ENMC international workshop: Limb girdle muscular dystrophies — Nomenclature and reformed classification Naarden, the Netherlands, 17–19 March 2017. Neuromuscular Disorders. 2018. № 28. Р. 702-710.
14. Влодавец Д.В., Казаков Д.О. Диагностические возможности магнитно-резонансной томографии мышц при нервно-мышечных заболеваниях. Неврологический журнал. 2014. № 3. С. 4-12.
15. Carlier P.G., Marty B., Scheidegger O., de Sousa P.L., Baudin P.-Y., Snezhko E., Vlodavets D. Роль количественной магнитно-резонансной томографии и спектроскопии скелетных мышц в оценке результатов клинических исследований (часть I). Нервно-мышечные болезни. 2016. № 4. С. 10-20.
16. Carlier P.G., Marty B., Scheidegger O., de Sousa P.L., Baudin P.-Y., Snezhko E., Vlodavets D. Роль количественной магнитно-резонансной томографии и спектроскопии скелетных мышц в оценке результатов клинических исследований (часть II). Нервно-мышечные болезни. 2017. № 1. С. 11-29.
17. Tasca G., Monforte M., Iannaccone E., Laschena F., Ottaviani P., Silvestri G., Masciullod M., Mirabellab M., Servideib S., Ricci E. Muscle MRI in female carriers of dystrophinopathy. European Journal of Neurology. 2012. № 19(9). Р. 1256-1260.
18. Lovitt S., Moore S.L., Franklin A. Marden F.A. The use of MRI in the evaluation of myopathy. Clinical Neurophysiology. 2006. № 117. Р. 486-495.
19. Мercuri E., Clements E. et al. Muscle magnetic resonanceimaging involvement in muscular dystrophies with rigidity of the spine. Ann. Neurol. 2010. № 67(2). Р. 201-208.

/23.jpg)
/23_2.jpg)